两年没看论文,实在难以专注,写笔记更有劲。
文章里的大部分术语都能在cs231n筆記里找到解释。
药物发现里一个主要的挑战是,设计具有所需属性的药物。化学空间里有成药潜力的分子有 10^23 到 10^60 个,人类已合成过的分子量级是 10^8 。研发一种新药的平均开销 1~2 billion 美元,在 100 亿人民币上下,平均研发时间是 13 年。
新药研发开销:
- 药物发现阶段 (Discovery Phase)
- 靶点筛选、化合物筛选、药理研究等;
- 成本占研发费用 10%~20%。
- 临床前研究 (Preclinical Phase)
- 动物实验、药物代谢研究、毒性试验等;
- 需要 1 到 3 年时间,成本在几千万美元到 1 亿美元之间。
- 临床试验阶段 (Clinical Trials)
- Phase I: 一期关注药物安全性,开销几百万到几千万美元在少数健康志愿者中进行;
- Phase II: 二期评估有效性和进一步安全性,开销几千万美元在少数患者中测试;
- Phase III: 三期是整个研发过程中最贵的阶段,要在成千上万患者中进一步证明有效性和安全性。
- 审批和上市阶段 (Approval and Launch Phase)
- 监管机构审批,提交大量的临床数据和文献,需要大量行政支持。
- 美国食品药物监管局 FDA (Food and Drug Administration) , 欧盟附属机构欧洲药品管理局 EMA (European Medicines Agency), 国家药品监督管理局 NMPA (National Medical Products Administration)
- 上市后市场推广和营销,需要上亿美元。
- 监管机构审批,提交大量的临床数据和文献,需要大量行政支持。
- 研发失败
- 药物研发的成功率非常低,只有1/5,000到1/10,000的候选化合物最终能够成功上市。
- 平均研发十三年和平均开销 1~2 billion 美元里面,有很大一部分项目在临床前和临床阶段失败。
传统上,化学家和药理学家通过他们的直觉和专业知识来识别新分子。尽管用 Lipinski’s RO5 可以减少可能成药的分子数量,但搜索空间还是过大。为了进一步减小搜索空间,人们开始使用 high-throughput screening (HTS) 高通量筛选,但任务仍然难以完成。现在,计算方法可以用于减小药物搜索空间和缩短研发新药的时间。
高通量筛选:
- 研究人员准备化合物库 (Compound Library) 或药物库 (Drug Library)
- 化学合成
- 可以通过多步合成,每步通过化学反应加上一个新的基团。
- 现代化学合成可以用自动化的反应装置,短时间合成大量化合物。
- 天然产物,即从植物、微生物、海洋生物等中,提取分离。
- 生物制药,从动物、人体提取。
- 药物衍生再设计,改造现有药物,通过增删改基团可以改变分子的性质。
- 基因工程微生物可以被设计成发酵过程中生产不同化学物质。
- 以上过程也可以跟计算机辅助药物设计 (CADD) 结合,用分子对接、分子动力学筛选化合物。
- 化学合成
- 实验筛选
- 在微孔板(通常是96孔、384孔、1536孔板等)的每个孔中放入一个化合物,
- 测量其与目标的相互作用,例如酶的活性变化、细胞生长或死亡、荧光信号变化等。
- 数据分析
property generation
3 个数据集:
- Moses 1.6M
- Zinc 250K
- GDB13 rand 1M
7 个 baseline:
- GCPN: graphs
- JTVAE: graphs
- MolGPT: SMILES
- MolT5?效果如何
- MolGAN: graphs
- MolDQN: graphs, Q-learning
- GraphDF: flow
- LSTM+RL
用这些数据的开源代码,在 QED 上优化,用 baseline 在各自论文里描述的 reward function。
Hardware: one TITAN RTX GPU, 60GB RAM, and an eight-core CPU.
模型超参:
- 4 masked transformer decoder layers
- 8 attention heads
- 1024d fc
- 512d embedding
- 3 epochs gpt
- 100 steps with each step averaging 500 molecules in RL stage
- REINFORCE algorithm discount factor 0.99, max seq size 150 chars
- character-level tokenization
其中 reward function:
Metrics:
- validity, rdkit 能生成 molecule 对象就是 1,否则 0;
- novelty, 训练集没出现的生成数据;
- diversity, 去重,这三个指标筛选后的再参与下面两个指标的比较。
- QED, 博客里聊过,这里用 8 个性质 (molecular weight, AlogP, hydrogen bond donors, hydrogen donor acceptors, molecular polar surface area, rotatable bonds, aromatic rings, structural alerts) 得到 0~1 结果;
- SAS, 大于 6 难以合成。
property optimization
2 个数据集:
- ChEMBL IC50,移除了只有范围数据的分子,只用治疗阿尔茨海默病的潜在靶点 BACE 1 这个 β-分泌酶1 (Beta-secretase 1) 的数据,从10164 条里筛选出了 9331 个样本。
- 400 个分子来自 anti-cancer activity dataset like FDA approval, clinical trials, DrugBank, etc
他们用 Chemprop 而不是 RDKit 预测分子的 anti-cancer 性质数据,用 batch-size 50 训练了 30 epochs, lr 1e-4, ensemble size 2.
然后对 ChEMBL 数据用 exp(pIC50/3) 为奖励函数,对抗癌数据用 Chemprop 预测的 0~1 probability 结果作为奖励函数。
得到的结果有问题:
- 3 个数据集的 Baseline 跟上面 Taiga stage 1 的数据对不上;
- 用 pIC50 作为奖励函数,模型评估指标也是 pIC50,这种 IC50 亲和力表现提高 20% 的提升是不是不太公平?
之后还有很多试验,包括 tokenizer 和 w/o w/ RL 的 Validity(↑) Divesity(↑) Novelty(↑) QED(↑) SAS(↓) 结果,但不知为何后者没有 SAS 结果。
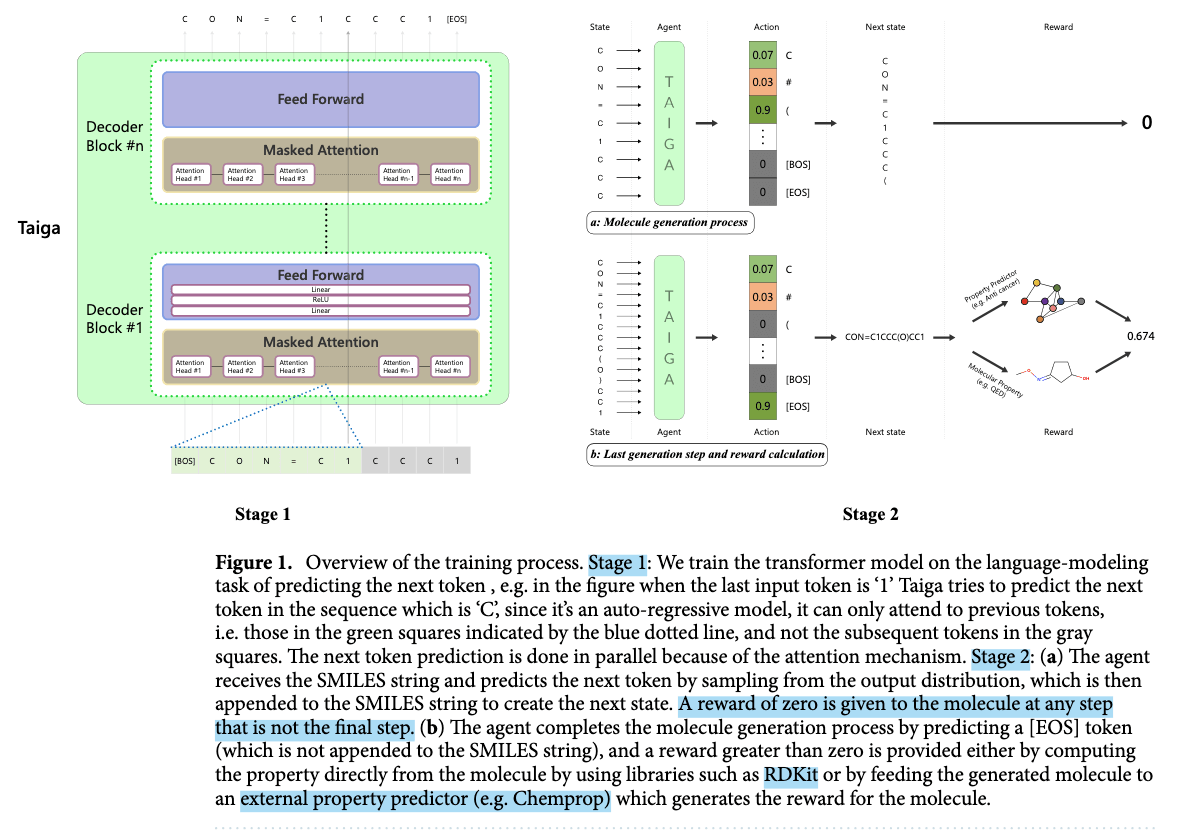
MDP: M = (S, A, P, R, γ ),这里我感觉文章里的表达逻辑混乱,梳理一下:
- 状态空间 $S = \{s_i\}$,$0 < i \leq t$,SMILES 串的长度 $t \leq T$ ,其中 $T$ 表示在模型生成 [EOS] 标记或达到最大长度时的终止状态;而 $s_0$ 是初始状态,它是一个空字符串。
- 每一个 $s$ 都被表示为一个向量?向量里每一个字符都来自 $\{0, \dots, n\}$ 的 SMILES 字符集。
- 动作空间 $A = \{a_i\}$,每个 $a_i$ 都来自 $\{0, \dots, n\}$ 的 SMILES vocabulary。
- 转移概率 P,由于状态空间和动作空间仅由字符组成,因此转移概率简单地为 $p(s_{t+1} | s_t, a_t) = 1$,因为添加字符是确定性的。
- 奖励函数 R,定义所有中间状态的奖励为零,即 $R(s_t) = 0$。$R(s_T) = f(s_T)$ 函数应用于最终生成的分子,$\gamma$ 是折扣因子。
具体做法是 stage 2 每一个 token 作为 action,最后如果符合要求(QED, anti-cancer)则 reward = 1,前面每一步都是 0。
为什么不合并为 1 个 stage?
大概因为刚开始训练时模型生成的 SMILES 太随机,符合要求的几率太低,都是无效样本。