在文獻裡 Molecular Generation 和 Molecule Generation 這倆說法各占一半。
Atom vs. Molecule
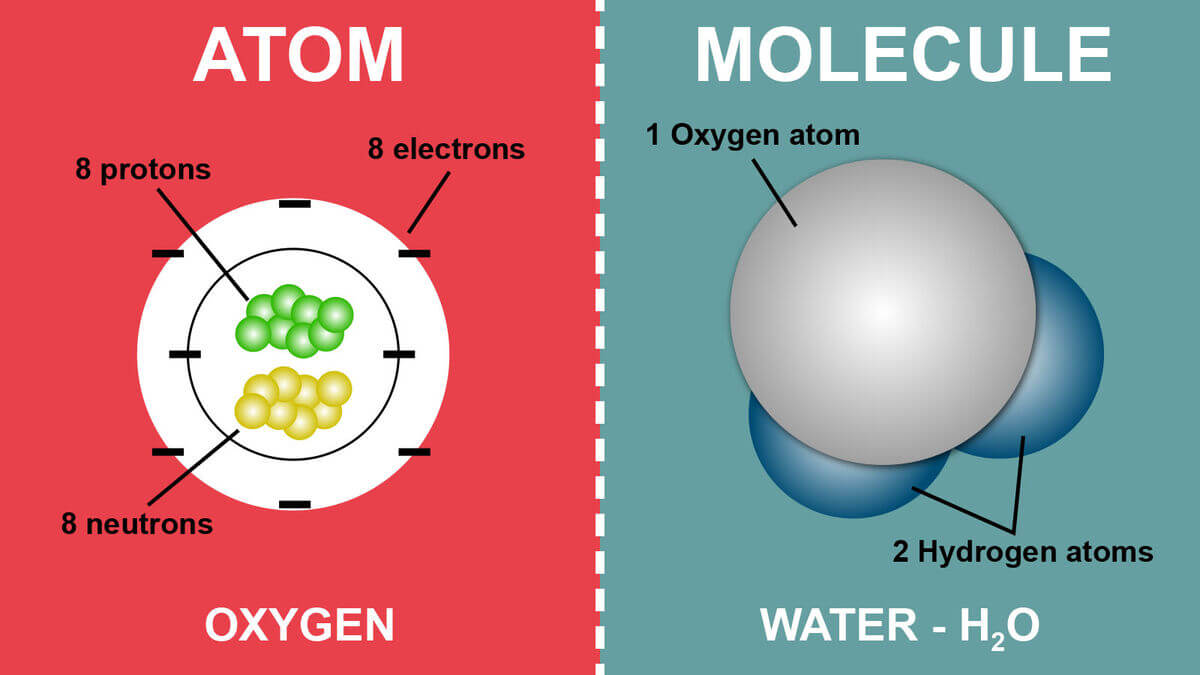
這裡的原子軌道其實是錯的,
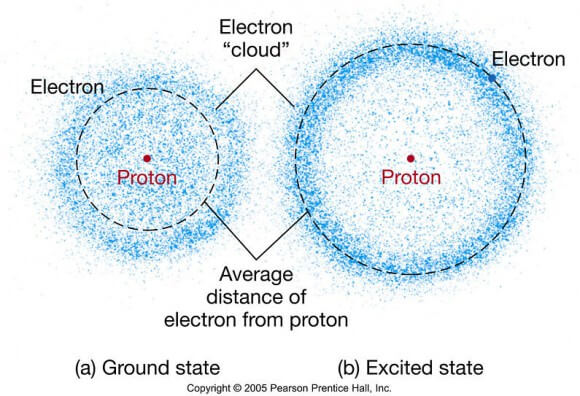
電子不是以確定的軌道運動的,是以波動的形式分布在原子核周圍,無法被精確定位,它的分布區域叫電子雲 (Electron Cloud) ,電子雲是一個概率分佈。
基態到激發態電子的能級變高,離原子核就越遠,改變軌道後會產生不同形狀的電子雲。
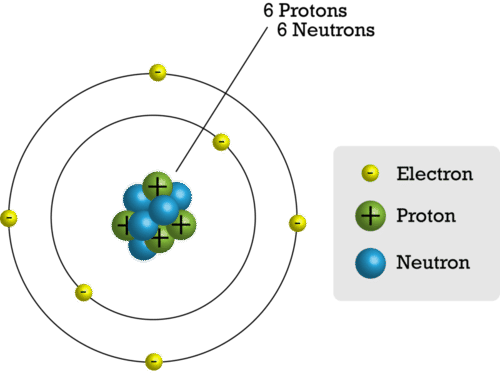
比如碳 Carbon C 的基態電子分布是1s² 2s² 2p²,其中第二能層有兩種軌道, p 軌道離原子核比 s 軌道更遠,所以能量更高,吸收能量到激發態時可能電子會先從 s 軌道跳到 p 軌道,這裡的吸收能量通常是光子能量,不同波長的光子對應不同的能量。激發態的 C 可能變成 1s² 2s¹ 2p³ / 1s² 2s² 2p¹ 3s¹ / 1s² 2s² 2p¹ 3p¹ 等。
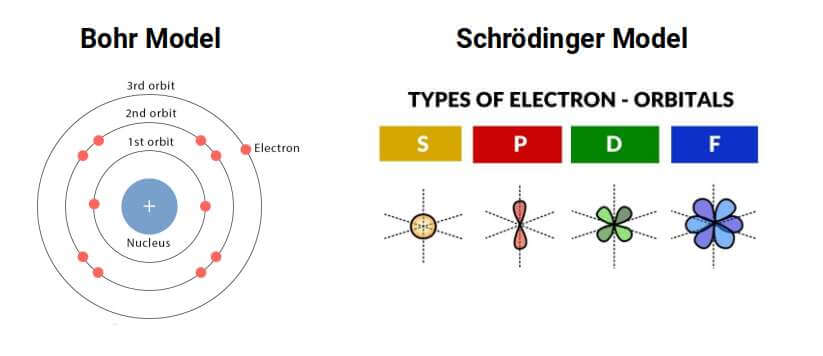
分子裡的電子跟原子類似,電子只能佔據有特定能量的分子軌道,分子軌道是各個原子的原子軌道通過量子力學的線性組合,化學鍵(共價鍵、離子鍵、金屬鍵)是原子的電子雲重疊的結果,是通過量子力學中的波動函數互動產生的量子效應。
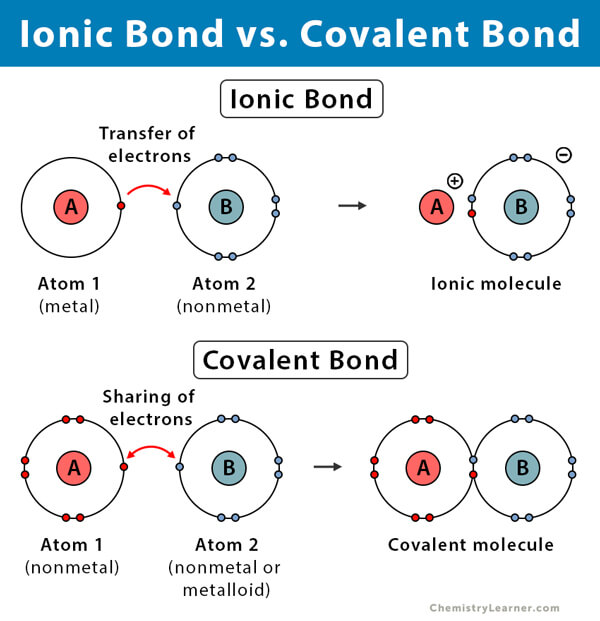
- 共價鍵 (Covalent Bond) 是兩個原子共享電子,比如乙炔的 C≡C 碳碳三重鍵和碳氫單鍵都是共價鍵。比如這裡的 C-H 碳氫單鍵是由 C 的 sp 混合軌道和 H 的 1s 軌道重疊所形成的較強 σ 鍵。
- 離子鍵 (Ionic Bond) 是一個原子將電子完全轉移到另一個原子,從而形成帶正電的陽離子和帶負電的陰離子。比如氯化鈉是 Na 原子將一個電子轉移給 Cl 原子形成 Na⁺陽離子和 Cl⁻陰離子。通常是活潑的金屬原子和非金屬原子形成,HCl是共價鍵。
- 金属键 (Metallic Bond) 是金屬元素間共享自由移動的電子,形成電子海 (Electron Sea),自由地在整個金屬結構中移動。比如鋼鐵 Fe 中的鐵原子由金屬鍵結合,自由電子(“束縛電子”或“導電電子”)在金屬原子間流動,並且在金屬原子周圍形成一種電場,使得正離子(金屬原子核)保持緊密排列並且相互吸引。
物質
物質(Substance)是指具有一定質量和體積的物體,它由原子、分子或離子等基本單位組成。物質不都是分子,分子只是原子通過共價鍵結合成的粒子,比如 NaCl 是離子化合物。
- 元素 (Elements) 是由相同類型的原子組成的純物質,無法分解成更簡單的物質。
- 化合物 (Compounds) 是由兩種或更多種不同化学元素通過化學鍵結合而成的物質,具有固定的成分比例,且不同於其組成元素的性質。
- 混合物 (Mixtures) 是由兩種或多種不同物質(元素或化合物)物理結合而成,成分比例可以變化,且各成分保持其原有性質。
有机物
有机物 / 有机化合物 (organic compound/chemical) 是分子中包含碳-氢或碳-碳共价键的化合物,通常等价于烃,常有碳碳键。
在去年的 wiki 里有过相关学习。
分子間作用力
原子與原子之間的作用力,我們稱之為『化學鍵』(離子鍵、共價鍵、金屬鍵);而分子與分子間的作用力 (Intermolecular force : IMF; also secondary force),有『凡得瓦力(van der Waals forces)』、『氫鍵(Hydrogen Bonding)』以及 Ion-dipole forces / Ion-ion forces。
極性
極性 (Polarity) 是指分子內部電荷分布不對稱,導致分子呈現某種程度的正負極性。所以離子一般都有極性。
共價鍵也會有極性,比如水分子 (H₂O),O 比 H 更具電負性,所以水分子中氧原子帶負電,氫原子帶正電。極性分子因此具有了偶極 (dipole),偶指的是分子內部有兩個電極(正、負)。
範德華力
是三種 Dipole-dipole force/interaction 偶極-偶極力。
相似相容 (like dissolves like)
溶解的本質是溶劑 (Solvent) 和溶質 (Solute) 之間的作用力大於溶劑分子之間、溶質分子之間的作用力,以結合溶質分子和溶劑分子。
溶解分為四種情況:
- 極性溶劑,極性溶質;
- 極性分子內部有偶極,極性分子之間偶極可以正負互相吸引,即普通的偶極-偶極力。
- 極性離子跟極性分子會產生 離子-偶極力。
- 非極性溶劑,非極性溶質;
- 是另一種範德華力,非極性文子沒有永久偶極,但有瞬時偶極(伦敦色散力)。
- 極性溶劑,非極性溶質;
- 溶解是一个克服能量障碍的过程。
- 極性溶液裡,溶劑分子之間的 偶極-偶極力 很強,溶質分子的瞬時偶極跟溶劑分子之間的 伦敦色散力 不足以破壞溶劑分子之間的力。
- 非極性溶劑,極性溶質,跟極性溶液非極性溶質一樣的道理。
一般來說溶劑都是 共價鍵 化合物,也有離子溶劑存在,如熔融鹽 (如 NaCl )。
離子-偶極力
Ion-dipole Forces
這是帶電的離子與極性分子之間的作用力。例如,在水溶液中,帶正電的離子(如Na⁺)會與水分子的負極性(氧端)相互作用,形成離子-偶極力。
離子-離子力
Ion-ion Forces
帶電離子之間的 靜電引力(庫倫力)$F = k_e \frac{|q_1 q_2|}{r^2}$,這個分子間作用力跟原子間的離子鍵一樣是因為靜電力粒子間相互吸引。
氫鍵
Hydrogen Bonding
氫鍵是一種特殊的分子間作用力,發生在氫原子與某些具有高電負性的原子(如氟、氧或氮)之間。氫鍵比一般的凡得瓦力強,並且在水(H₂O)、氟化氫(HF)、氨(NH₃)等分子中起著重要作用。
例如,在水分子中,氫原子與氧原子之間形成氫鍵,使得水的熔點和沸點比許多其他小分子要高。
結構和性質對應關係
| 特性 | 共價鍵(Covalent Bond) | 離子鍵(Ionic Bond) | 金屬鍵(Metallic Bond) |
|---|---|---|---|
| 導電性 | 通常是非導電的,因為它們不容易離解成自由的離子。 | 通常是良好的導電體,當它們溶解在水中或熔化時,能形成自由移動的離子,導電性顯著。 | 自由電子可以在金屬內自由移動,使金屬能夠良好導電。 |
| 熔點和沸點 | 通常較低,因分子之間的相互作用較弱。 | 通常較高,因為強大的靜電吸引力需要較多的能量來克服。 | 通常較高,金屬鍵的強度較大,需要較大能量來克服金屬原子間的吸引力。 |
| 溶解性 | 分子极性较强或能形成氢键,就能溶于水,否則溶於非極性溶劑。 | 通常容易溶解在水等極性溶劑中,形成帶電的離子。 | 大多數金屬不易溶解於水或一般溶劑中,但可以在其他金屬中形成合金。 |
| 結構 | 通常具有分子結構,分子之間的相互作用較弱(如氫鍵)。 | 通常形成晶體結構,由離子組成,離子之間通過靜電吸引力結合。 | 具有金屬晶格結構,原子通過金屬鍵保持在一起。 |
| 延展性和可塑性 | 通常較脆,容易破裂。 | 通常較脆,受外力作用時容易斷裂。 | 具有良好的延展性和可塑性,原子能在外力作用下滑動而不破裂。 |
| 光澤 | 通常無光澤,部分共價化合物可能呈現不同的顏色。 | 通常無光澤,但在某些情況下可能具有光澤(例如鹽晶體)。 | 金屬具有光澤,金屬表面能反射光線,產生金屬光澤。 |
一些性質參數
LogP(疏水度)
P 是分子在 辛醇(octanol)和水之间的分配系数 (Partition Coefficient)。(octanol-water partition coefficient)
$P = \frac{[\text{分子在辛醇中的浓度}]}{[\text{分子在水中的浓度}]}$
$\text{LogP} = \log_{10}(P)$
- LogP > 0 表示親脂,因為在辛醇裡濃度比水中大。
- LogP < 0 表示親水,因為在辛醇裡濃度比水中小。
LogP = 0,親水性 = 疏水性。
药物设计:LogP 影响药物的 吸收、分布和渗透性,通常要求 LogP 值适中(例如 0-5)。
- Lipinski’s Rule of Five 中,LogP < 5 是成药性的重要规则之一。
- 苯(C6H6):LogP ≈ 2.1(疏水性较强);乙醇(CH3CH2OH):LogP ≈ -0.2(亲水性)。
LogS (親水度)
S 是分子在水中的溶解度 (Solubility)。
$S = 分子在水中的饱和溶解度(mol/L)$
- 药物开发:良好的溶解性(高 LogS)对药物吸收和生物利用度至关重要。
- 乙醇:LogS ≈ 1.6(溶解性高)。
- 阿司匹林:LogS ≈ -2.0(溶解性较低,需要助溶策略)。
MW
Molecular Weight: 分子质量,单位道尔顿 (dalton, Da) 是碳 12 (质子和中子数都是 6 ) 质量的 $\frac{1}{12}$ 即 $= 1.661×10^{-27} kg$。
比如在药物设计里,过大分子可能难以通过细胞膜。
TPSA
Topological Polar Surface Area: 分子上所有极性原子的表面积,常用于评估分子的透过生物膜的能力。
Hydrogen Bond Donors and Acceptors, RO5
氢键供体和受体的数量,影响分子的生物活性。
Lipinski’s rule of five:
- No more than 5 hydrogen bond donors (the total number of nitrogen–hydrogen and oxygen–hydrogen bonds)
- No more than 10 hydrogen bond acceptors (all nitrogen or oxygen atoms)
- A molecular mass less than 500 daltons
- A calculated octanol-water partition coefficient (Clog P) that does not exceed 5
藥物設計的五個基本規則,rule of five(RO5),包括上面說到的logP、分子質量、氫鍵供受體。
導電性
- HOMO-LUMO Gap (Highest Occupied Molecular Orbital - Lowest Unoccupied Molecular Orbital Gap):
- HOMO-LUMO间隙是指分子的最高占据分子轨道(HOMO)与最低空分子轨道(LUMO)之间的能量差。
- homo-lumo gap 越小,分子中的电子越容易从HOMO跃迁到LUMO,从而激发出电荷载体,这有助于电子的流动,提高导电性。
- 金属材料(如金、银)的HOMO-LUMO间隙几乎为零,意味着电子能在材料中自由流动,因此它们具有非常好的导电性。
- 有机半导体材料(如有机太阳能电池中的材料)通常具有适中的HOMO-LUMO间隙,使其在特定条件下能够导电。
- 绝缘材料(如陶瓷或某些高分子材料)具有较大的HOMO-LUMO间隙,因此它们几乎不导电。
- Electron Affinity:
- 電子親和力表示分子或材料获取一个电子的能力。通常以电子伏特(eV)为单位。
- 親和力越高,材料的导电性越好。
- 有机半导体材料(如聚合物电池或有机光伏材料)通常具有较高的电子亲和力,允许电子在电场作用下迅速移动,从而提高导电性。
- Charge Mobility:
- 電荷遷移率表示电荷载体(如电子或空穴)在电场作用下的迁移速度。
- 迁移率越高,材料的导电性越好。
- Polarizability:
- 极化性是指材料在外电场作用下电子云的变形程度,极化性高的材料能更好地响应外电场。
- 高极化性的材料能更好地调节其电子云,增加电子流动性。
- 太陽能電池的有機材料聚苯胺(PANI)和聚吡咯(PPy)有很高極化性。
- Ionization Energy:
- 电离能是将分子中的一个电子从其最低能级移除所需要的能量。
- 低电离能的材料更容易释放电子,釋放電子後材料會表現強導電性。
- 电金属如铜和银有較低的电离能。
- Electron Density:
- 分子的电子密度高,电子就容易在分子内自由移动。
- 导电金属如铜和银有非常高的电子密度。
pKa, pKb
pKa (Acid dissociation constant): 用来描述酸在水溶液中的离解程度,是分子酸性或碱性强弱的一个重要参数。
强酸:$pKa$ 值较小,酸容易离解;弱酸:$pKa$ 值较大,酸不容易离解。
用 pKa 能得到 pKb (Base Dissociation Constant, Kb)。
pK
pK 是亲和力值的负对数,即 pK = -log(亲和力)。
亲和力
什么是配体——
配体 (ligand) 可以理解为一种“钥匙”,它能够与蛋白质或其他大分子上的“锁”(即靶标, target)相结合。这个“锁”通常是蛋白质表面上的一个特定部位,也叫做结合位点。当配体(钥匙)与锁结合时,它会改变锁的功能或启动某些生物反应。
比如药物分子就是配体,它们通过结合到病菌或人体内某些蛋白质上,来调节这些蛋白质的活性,从而达到治疗疾病的效果。配体不仅仅是药物,也可以是体内的激素、神经递质等分子,它们都在调节身体各种功能时发挥着重要作用。
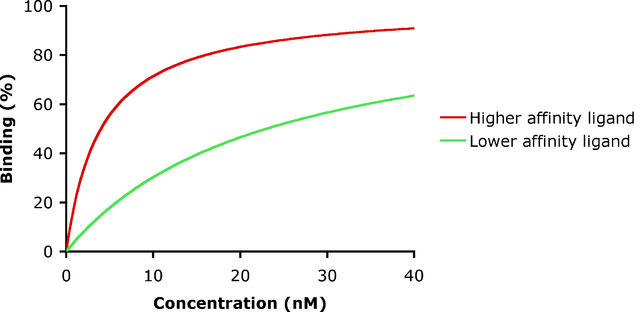
AutoDock 中的“Dock”即所谓 Molecular Docking 分子对接是指预测 / 模拟配体小分子如何跟靶标大分子结合,预测结合模式、结合强度以及生物学效果,比如模拟阿司匹林分子在COX酶的结合位点中的各种可能的构象,评估每种构象的结合强度。
分子之间 / 抗原抗体亲和力检测方法有——
- 动力学方法
- 表面等离子共振 (SPR),
- 生物膜干涉 (BLI),
- 热力学方法
- 等温滴定量热法 (ITC)
- 差示扫描量热仪 (DSC)
- 饱和浓度法
- ELISA
指标有——
- IC50 半抑制浓度(或称 IC₅₀ 为半抑制率,半最大抑制浓度)
- 描述一个化合物抑制目标(如酶或受体)功能的浓度。表示实验中抑制作用达到50%的化合物浓度。
- 单位是 μM 或 nM 表示药物的浓度。
- IC50 低于 100 nM 被认为有高亲和力,低于 10 nM 意味着分子对目标亲和力非常强,高于 100 nM 甚至超过 1 μM 表示亲和力弱。
- pIC50=-log(IC50),p 只有在化学里表示 negative logarithm,比如 pH = -log₁₀([H⁺] mol/L)。
- EC50
- Ki
- 抑制常数(inhibition constant),反应抑制剂对靶标的抑制强度,越小越强。
- $Ki=\frac{IC50}{1+\frac{[S]}{Km}}$ S是底物浓度;
- $Ki=\frac{IC50}{1+\frac{[A]}{EC50}}$ A是受体激动剂浓度
- Kd、KD 一回事
- 解离常数(dissociation constant)
- Ka
- 结合常数(association constant)
- Ka=1/Kd
- Km
- 米氏常数(Michaelis-Menten constant)
- 酶本身的一种特征参数,酶促反应达到最大反应速度一半时底物S的浓度。
- Kon
- 结合速率常数(association rate constant)
- 代表分子间结合时的快慢,单位为$M^{-1}∙S^{-1}$。
- Koff
- 解离速率常数(dissociation rate constant)
- 代表分子间解离时的快慢,单位为$S^{-1}$。
分子生成
- Target-Agnostic Generation
- 目标无关生成,類似 vanilla vae 和 gan 都是不加控制的、没有特定目标的生成。
- Target-Aware Generation
- 目标相关生成,在训练时把目标信息作为输入,或在奖励函数中引入目标信息,類似 conditional vae/gan,允許用類別標籤、圖像、文本描述控制生成。
- Conformation Generation
- 构象生成,考慮分子三維結構(即構象),這對性質也至關重要,构象生成不仅需要生成分子的二维连接图,还需要考虑分子的空间排列(如旋转、弯曲等)。
Data
雖然說物質(元素或化合物)不都是分子,但分子生成的數據是廣義的分子,比如離子化合物和通常以晶體結構存在的純金屬也算。
- 1D 分子表示方法,除了化学计量式 Stoichiometric formula 如水(H₂O)二氧化碳(CO₂)之外有以下几种,
- SMILES
- 以及各种变种,比如规范化 canonical SMILES,通过把 SMILES 展开为图 graph 结构,然后字典排序 (Lexicographic Order),从最小原子以唯一的遍历顺序便利环和分支结构,确保每个分子有唯一的 SMILES 表示。
- InChI
- Morgan Fingerprints
- SMILES
2D,由于数据比较少,直接在这列举,
Pixels (像素)
USPTO-30K,10K 行数据,300M;
DECIMER,手绘分子图,5K 张
Molecule OCR Real images Dataset,分子截图,300 张
- 3D datasets
- Voxels (体素,体积元素)
这些表示方法的唯一性和可翻转性见附录。
SMILES
Simplified Molecular Input Line Entry System
| 语法元素 | 描述 | 示例 |
|---|---|---|
| 原子 | 元素符号表示原子 | C (碳), O (氧), N (氮) |
| 单键 | 默认连接方式,无需显示 | C-C(乙烷) |
| 双键 | 用 = 表示 |
C=C(乙烯) |
| 三键 | 用 # 表示 |
C#C(乙炔) |
| 环结构 | 使用数字表示环中的连接位置 | C1CCCCC1(环己烷) |
| 分支 | 使用括号表示分支结构 | CC(C)C(异丙烷) |
| 芳香环 | 小写字母表示芳香性环结构 | c1ccccc1(苯) |
| 电荷 | 使用 + 或 - 表示电荷 |
[Na+](钠离子) |
| 氢原子 | 默认省略,若显示可通过 H 或 H# 表示 |
CH4(甲烷) |
| 立体化学 | 用 @ 或 @@ 表示立体化学配置 |
C[C@H](O)C(立体异构) |
| 官能团 | 使用合适的符号表示,如羧基 C(=O)O |
CC(=O)O(乙酸) |
| 多分子 | 用 . 表示多个分子 |
C1CC1.C1CC1(两个环己烷分子) |
| 键连接 | 用 - 或 = 连接多个分子 |
C-C(乙烷) |
| 分子中的其他原子 | 省略不常见的原子和其氢原子,如 O、N 可不标明氢 |
C=O(醛基) |
| 例子 | SMILES | 描述 |
|---|---|---|
| 甲烷 | CH4 |
四氢化碳 |
| 乙烯 | C=C |
两个碳原子用双键连接 |
| 环己烷 | C1CCCCC1 |
一个六碳的环结构 |
| 苯 | c1ccccc1 |
芳香环结构(苯) |
| 乙酸 | CC(=O)O |
醋酸 |
| 甲醇 | CO |
一个碳与氧连结的分子 |
| 氯化钠 | [Na+].[Cl-] |
两个离子:钠和氯 |
| 异丙烷 | CC(C)C |
由甲基取代的丙烷 |
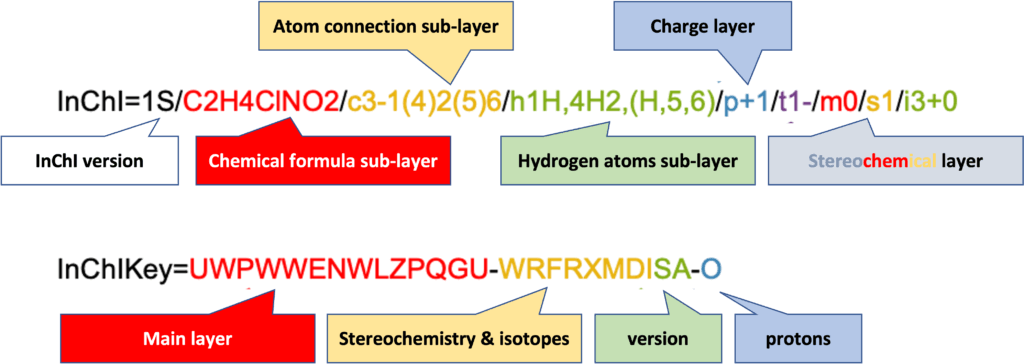
InChI
比如乙醇 (Ethanol):
InChI=1S/C2H6O/c1-2-3/h3H,2H2,1H3

- Main layer (always present)
- 1:版本號,目前是 version 1;
- S:standard,如果是 non-standard 就是 InChI=1/C2H6O/c1-2-3/h3H,2H2,1H3 略過 S;
- 分子式 (無前綴)
- C2H6O:由2个碳原子(C)、6个氢原子(H)和1个氧原子(O)组成;
- 分子的化学键 (前綴 “c” )
- c1-2-3:atom 1 与 atom 2 之间有一个单键,atom 2 与 atom 3 之间也有一个单键;
- 再举两个例子,C2H6 乙烷:InChI=1S/C2H6/c1-2/h1-2H3;
- InChI=1S/C2H4/c1-2/h1-2H2 乙烯和 InChI=1S/C2H2/c1-2/h1-2H 乙炔,化学键层都是 c1-2,是通过后面每个 atom 上 H 的数量体现二键 h1-2H2 三键 h1-2H。
- 氢的排列 (前綴 “h” )
- h3H,2H2,1H3:atom 3 (氧) 上有一个 H,atom 2 上有 2 个 H,atom 1 上有 3 个 H。
- 再举两个例子,C3H6 环丙烷:InChI=1S/C3H6/c1-2-3-1/h1-3H2,这里的 1-3H2 表示 C1 C2 C3 都有 2 个 H;
- C3H8 丙烷:InChI=1S/C3H8/c1-3-2/h3H2,1-2H3,对比上面 c1-2-3-1 和 h3H2,1-2H3。
- 其他层不会在所有 InChI 里出现,比如电荷层 “q” charge sublayer,质子层 “p” proton sublayer,也都是为了唯一标识一个分子而存在,见下图。
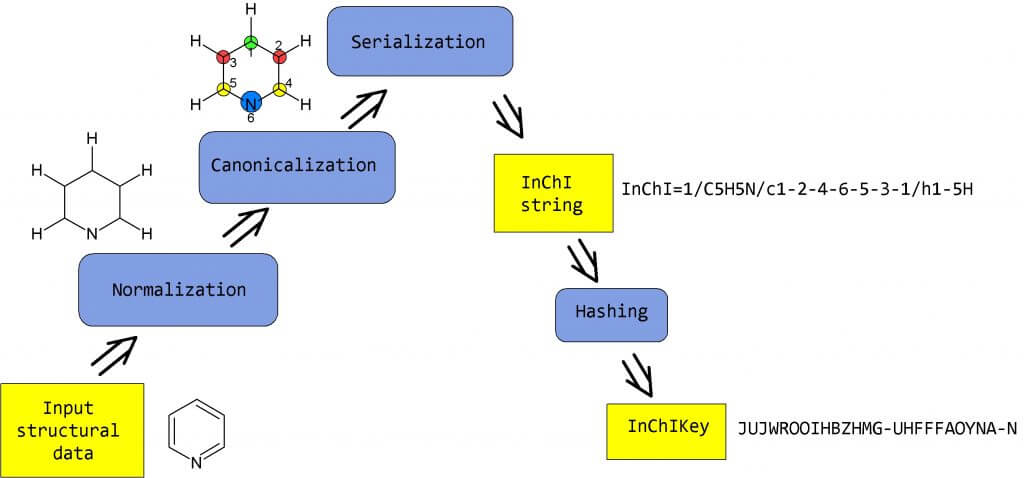
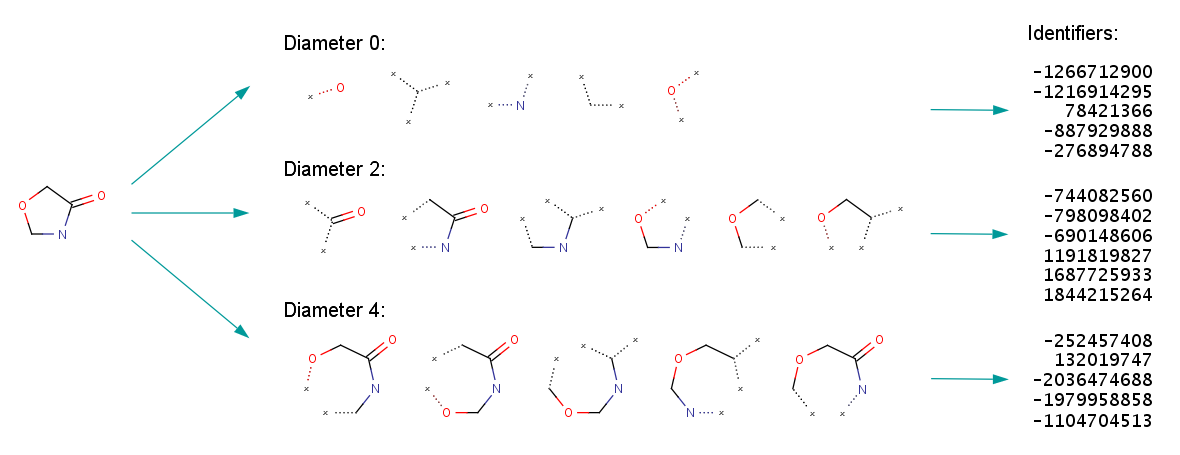
Morgan Fingerprints
以乙醇为例,
H H |
大致计算步骤是:
- 给 C1, C2, O, H1, H2, H3, H4, H5, H6 定义初始哈希值表示类型和邻域。
- C1 邻域:C2、H1、H2、H3;
- C2 邻域:C1、H4、H5、OH;
- O 邻域:C2、H6。
扩展邻域,把更多原子加入原先邻域。
扩展 iterations 中的所有邻域都生成对应哈希值。
对应填充 fingerprint,
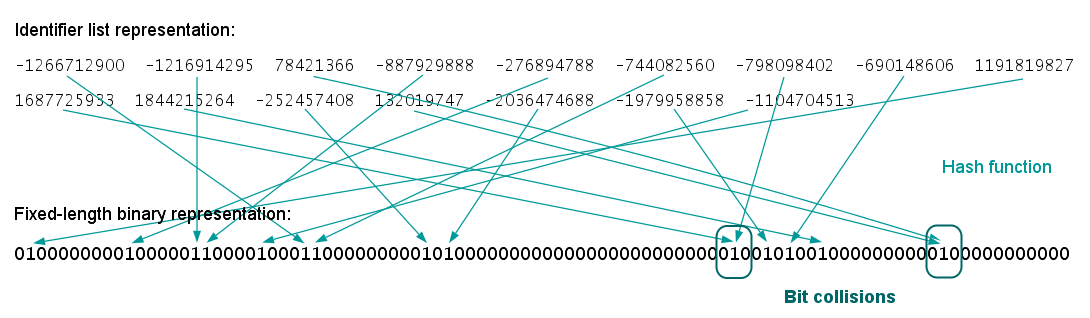
- 每个哈希值对应一个二进制位,即指纹中的每一位 1 对应存在此邻域,0 对应不存在此邻域;
- fingerprint 是固定长度的二进制码,如 1024 / 2048 等。
实际应用时会用 Morgan 原子邻域拓扑结构指纹算法的其他众多变种:
- ECFP (Extended Connectivity Fingerprints)
- FCFP (Functional Connectivity Fingerprints)
- MACCS Keys
- PubChem Fingerprints
…
1D/2D datasets
分别列举每个数据集的量级、主要构成、一条样本。
- GDB
- Generated Database
- GDB-11 enumerates small organic molecules up to 11 atoms of C, N, O and F following simple chemical stability and synthetic feasibility rules.
- GDB-13 enumerates small organic molecules up to 13 atoms of C, N, O, S and Cl following simple chemical stability and synthetic feasibility rules. 977 468 314 structures
- GDB-17 enumerates 166.4 billion molecules of up to 17 atoms of C, N, O, S, and halogens
- GDB-13 和 GDB-17 的前五條數據如下所示:
S1C=CC=C1
O1C=CC=C1
C1CCCC1
N1C=CC=C1
S1C=CC=N1
BrC1=C2C3=C4C(CC3CCC2=O)C(=N)NC4=N1
BrC1=C2C3C4CCC(C4)C3C(=N)OC2=NC=C1
BrC1=C2C3C4CCC(O4)C3(OC2=NC=C1)C#C
BrC1=C2C3C4CNC(C4)(C#N)C3OC2=NC=C1
BrC1=C2C3=C4C(OC(=O)C4=CC2=O)=CC3=NO1
QM9
- 全稱 Quantum Machine,paper。
- 是 133885 个来自 GDB-17 用简单化学稳定性和易合成规则生成的,最多 9 个 CONF 原子(不包括 H )构成的有机物数据集。
數據來源,sgdb9nsd_000001.xyz 第一条数据如下所示:
5
gdb 1 157.7118 157.70997 157.70699 0. 13.21 -0.3877 0.1171 0.5048 35.3641 0.044749 -40.47893 -40.476062 -40.475117 -40.498597 6.469
C -0.0126981359 1.0858041578 0.0080009958 -0.535689
H 0.002150416 -0.0060313176 0.0019761204 0.133921
H 1.0117308433 1.4637511618 0.0002765748 0.133922
H -0.540815069 1.4475266138 -0.8766437152 0.133923
H -0.5238136345 1.4379326443 0.9063972942 0.133923
1341.307 1341.3284 1341.365 1562.6731 1562.7453 3038.3205 3151.6034 3151.6788 3151.7078
C C
InChI=1S/CH4/h1H4 InChI=1S/CH4/h1H4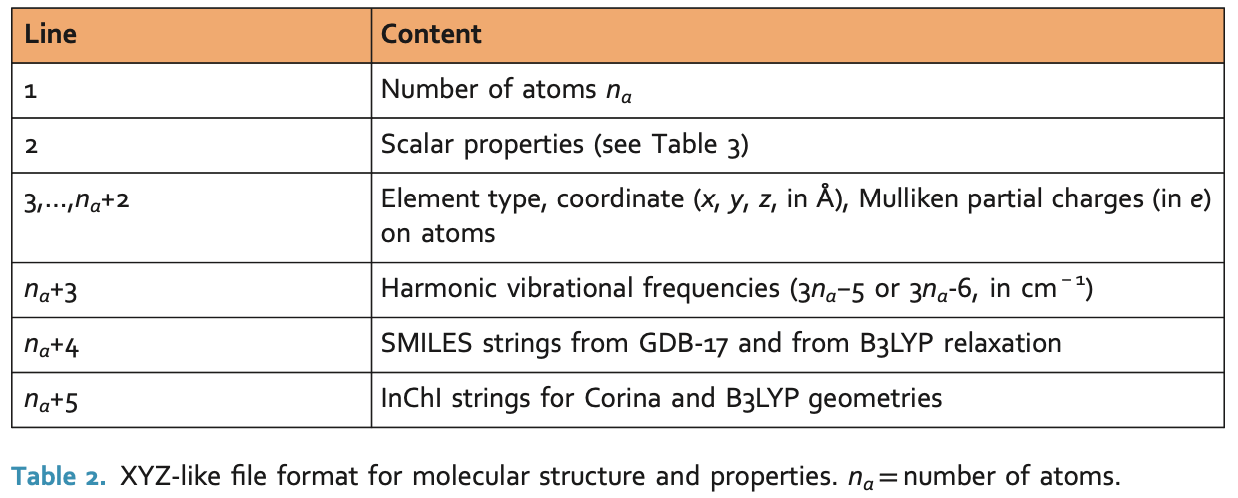
还有另一种形式,包含能量和距离矩阵 / 邻接矩阵,在 chainer 框架里有这个数据集。
ZINC-20
鋅-20,通过疏水度 logP 和分子质量 MW 划分 Tranches 提供数据下载。
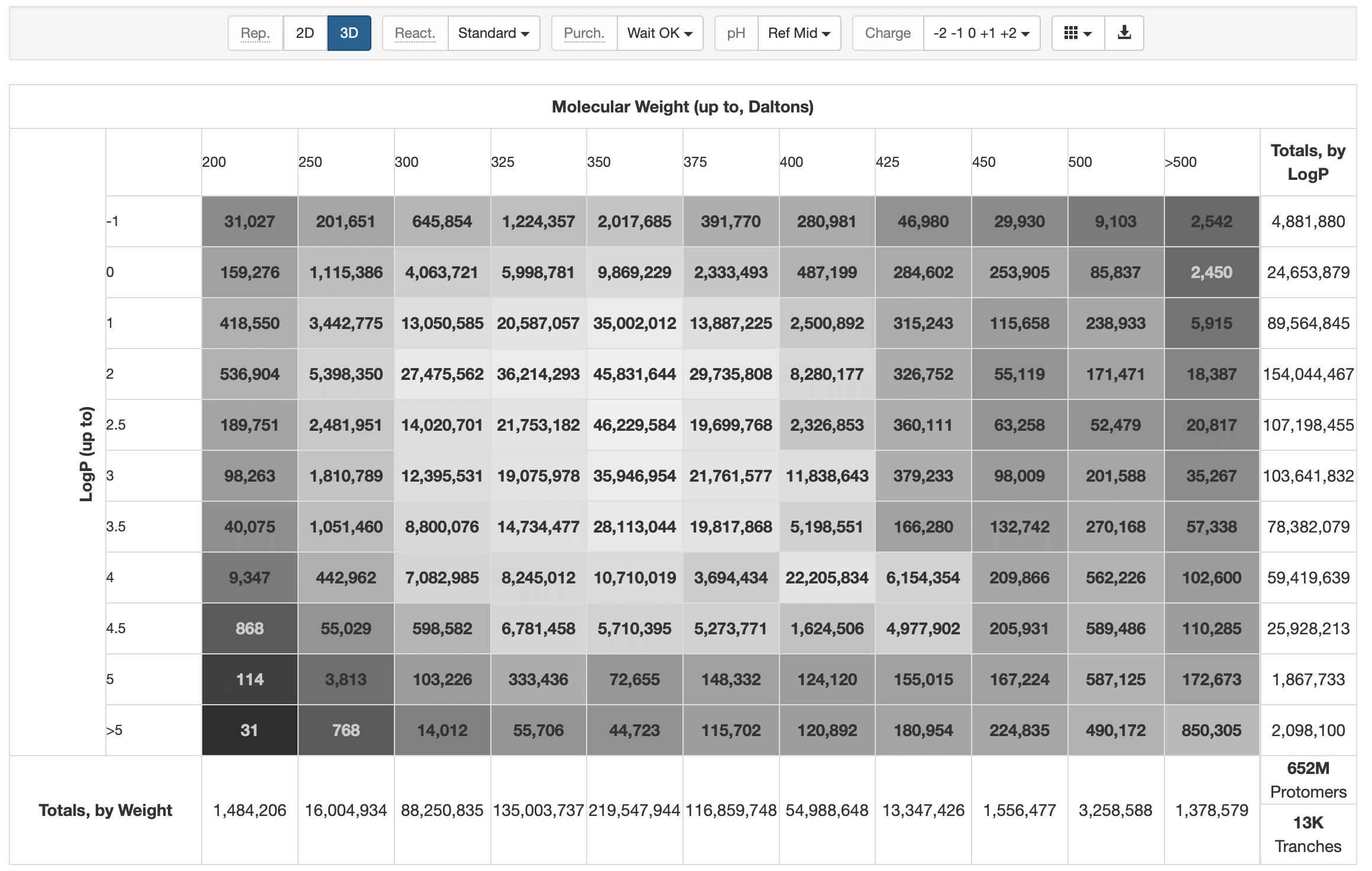
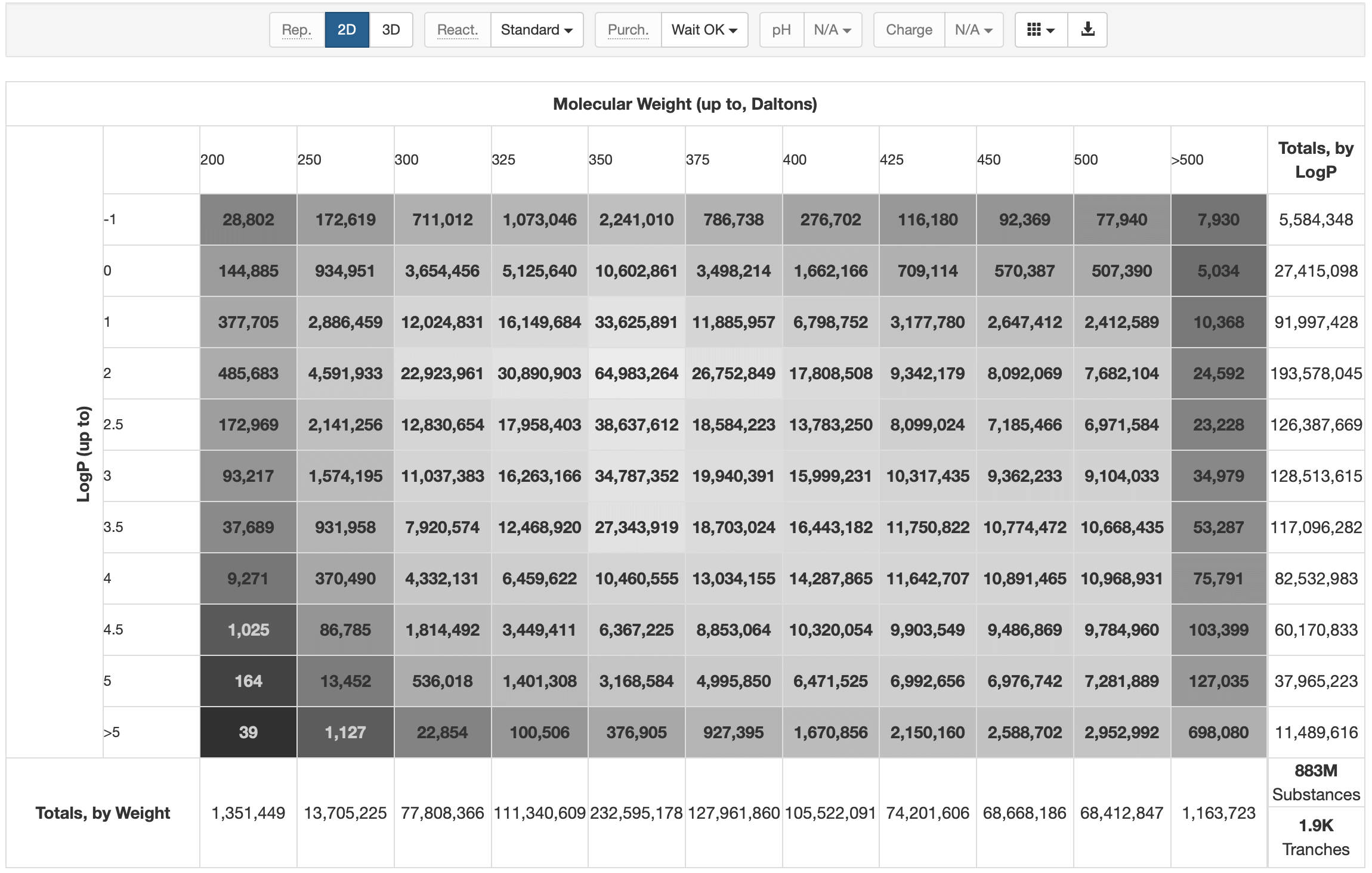
數據支持很多格式,包括 1D SMILES,2D 以 tab 间隔的 txt,3D 的 DOCK37.db2, AutoDock.pdbqt, Mol2.mol2, SDF.sdf。
- 除了从官网手动下载外,用 curl 或 wget 以 mol2 格式为例 tranches 的下载方式:
curl --remote-time --fail --create-dirs -o AA/AAML/AAAAML.xaa.mol2.gz http://files.docking.org/3D/AA/AAML/AAAAML.xaa.mol2.gz
curl --remote-time --fail --create-dirs -o AA/AAMM/AAAAMM.xaa.mol2.gz http://files.docking.org/3D/AA/AAMM/AAAAMM.xaa.mol2.gz
curl --remote-time --fail --create-dirs -o AA/AAMN/AAAAMN.xaa.mol2.gz http://files.docking.org/3D/AA/AAMN/AAAAMN.xaa.mol2.gz - 1D 和 2D 是 SMILES + ZINC ID + 一些性质参数,3D 会包含原子坐标、分子间作用力和电性等数据,以 AutoDock 格式为例:
MODEL 1
REMARK Name = ZINC000004228267
REMARK x y z vdW Elec q Type
REMARK _______ _______ _______ _____ _____ ______ ____
ROOT
ATOM 1 N UNK A 0 2.396 -0.224 1.517 0.00 0.00 -0.740 N
ATOM 2 H UNK A 0 2.098 0.687 1.372 0.00 0.00 +0.400 HD
ENDROOT
BRANCH 1 3
ATOM 3 C UNK A 0 3.745 -0.489 1.727 0.00 0.00 +0.230 A
- ChEMBL
- ChEMBL or ChEMBLdb是一个手动整理的化学数据库,包含具有药物诱导性质的生物活性分子。
- chEMBL 中的 EMBL 是 European Molecular Biology Laboratory,跟 ZINC 一样有很多种数据,如 2.5M compounds, 92.1k documents, 37.2k activities。
- 可用属性有38个条目,实际在可下载的官网化合物csv里有36条 ↓ ,表格见附录。
"ChEMBL ID";"Name";"Synonyms";"Type";"Max Phase";"Molecular Weight";"Targets";"Bioactivities";"AlogP";"Polar Surface Area";"HBA";"HBD";"#RO5 Violations";"#Rotatable Bonds";"Passes Ro3";"QED Weighted";"CX Acidic pKa";"CX Basic pKa";"CX LogP";"CX LogD";"Aromatic Rings";"Structure Type";"Inorganic Flag";"Heavy Atoms";"HBA (Lipinski)";"HBD (Lipinski)";"#RO5 Violations (Lipinski)";"Molecular Weight (Monoisotopic)";"Np Likeness Score";"Molecular Species";"Molecular Formula";"Smiles";"Inchi Key";"Inchi";"Withdrawn Flag";"Orphan"
CHEMBL1807862;"";"";"Small molecule";"";"279.72";"1";"1";"3.84";"22.00";"3";"0";"0";"1";"N";"0.66";"None";"None";"3.99";"3.99";"3";"MOL";"-1";"18";"2";"0";"0";"278.9921";"-1.36";"None";"C13H7ClFNOS";"O=c1c2cccc(F)c2sn1-c1ccc(Cl)cc1";"HENRYDPMUODAEA-UHFFFAOYSA-N";"InChI=1S/C13H7ClFNOS/c14-8-4-6-9(7-5-8)16-13(17)10-2-1-3-11(15)12(10)18-16/h1-7H";"False";"-1" - 在 assays 里有 1.7 M 条分子的很短的 description 可以用于 captioning,比如 959390 这个分子的描述是,“Cytotoxicity against human CCRF-CEM cells by SRB assay” 根据 SRB 分析此物质对人类 CCRF-CEM 有细胞毒性。
- CEPDB
- The Clean Energy Project Database, 2.3 million molecular motifs, derived from 150 million density functional theory calculations.
- 是 CEP Harvard Clean Energy Project 的一部分,2012 和 2013 版共15.14 GB(压缩后,解压的 cepdb_2012-10-24.sql 有 61 GB,建表),geometry 三维坐标数据共 18.54 GB。
- 关注能量相关属性,除了 HOMO-LUMO gap 之外,还有 Photovoltaic performance parameters 光伏表现参数 PCE: the power conversion efficiency Voc: the open-circuit voltage Jsc: the short-circuit current density。
--
-- Table structure for table `data_calcqcset1`
--
DROP TABLE IF EXISTS `data_calcqcset1`;
SET @saved_cs_client = @@character_set_client;
SET character_set_client = utf8;
CREATE TABLE `data_calcqcset1` (
`id` int(11) NOT NULL AUTO_INCREMENT,
`mol_geom_id` int(11) NOT NULL,
`calc_id_str` varchar(250) COLLATE utf8_bin NOT NULL,
`calc_tbz_str` varchar(250) COLLATE utf8_bin NOT NULL,
`calc_archive_subdir_path` varchar(250) COLLATE utf8_bin NOT NULL,
`modelchem_str` varchar(100) COLLATE utf8_bin NOT NULL,
`e_total` double DEFAULT NULL,
`e_homo_alpha` double DEFAULT NULL,
`e_lumo_alpha` double DEFAULT NULL,
`e_gap_alpha` double DEFAULT NULL,
`e_homo_beta` double DEFAULT NULL,
`e_lumo_beta` double DEFAULT NULL,
`e_gap_beta` double DEFAULT NULL,
`e_gap_min` double DEFAULT NULL,
`dipmom_total` double DEFAULT NULL,
`s2_val` double DEFAULT NULL,
PRIMARY KEY (`id`),
UNIQUE KEY `calc_id_str` (`calc_id_str`),
KEY `data_calcqcset1_2174315b` (`mol_geom_id`),
CONSTRAINT `mol_geom_id_refs_id_9f7beeea` FOREIGN KEY (`mol_geom_id`) REFERENCES `data_molgeom` (`id`)
) ENGINE=InnoDB AUTO_INCREMENT=113757770 DEFAULT CHARSET=utf8 COLLATE=utf8_bin;
SET character_set_client = @saved_cs_client;
--
-- Dumping data for table `data_calcqcset1`
--
-- ORDER BY: `id`
LOCK TABLES `data_calcqcset1` WRITE;
/*!40000 ALTER TABLE `data_calcqcset1` DISABLE KEYS */;
INSERT INTO `data_calcqcset1` VALUES (1,16855,'A.16.C13H8N2S.49.noopt.bp86.sto6g.n.sp','','','MM//BP86/STO-6G',-1004.9549135703,-0.101,0.019,0.12,-0.101,0.019,0.12,0.12,3.9718,NULL),(2,16855,'A.16.C 13H8N2S.49.noopt.bp86.svp.n.sp','','','MM//BP86/SVP',-1007.3925734934,-0.189,-0.073,0.116,-0.189,-0.073,0.116,0.116,2.9927,NULL),(3,15011810,'A.16.C13H8N2S.49.bp86.svp.n.bp86.svp.n.sp','','','BP86/S VP//BP86/SVP',-1007.4106491085,-0.189,-0.076,0.113,-0.189,-0.076,0.113,0.113,3.3101,NULL), -- vim 读大文件真好用,1000 行的 id 在 500 万左右,不知总量多少。
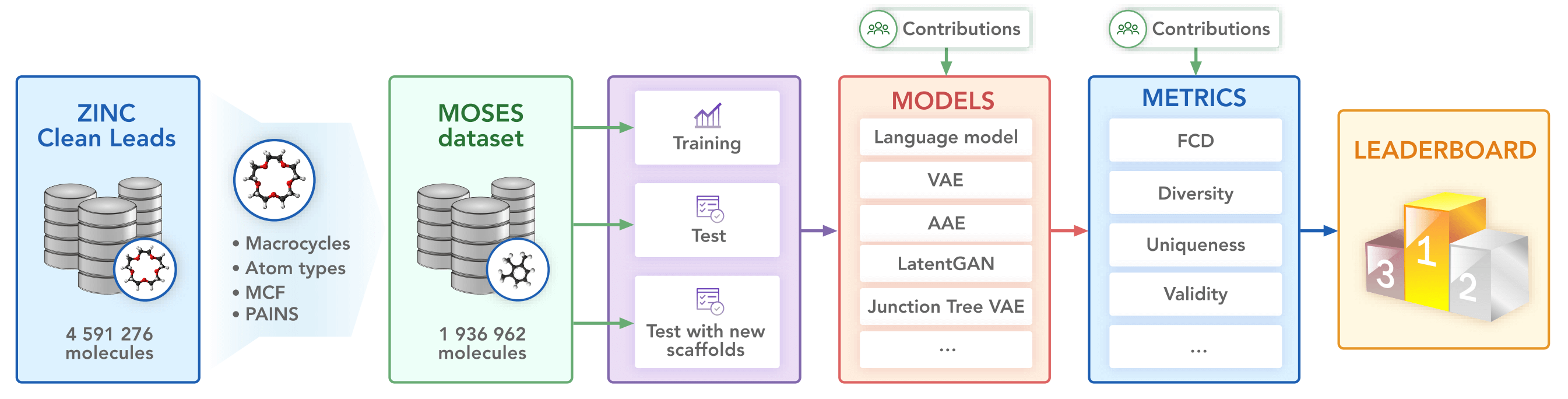
- MOSES
- Molecular Sets 来自 ZINC Clean Leads,分子量范围为 250 到 350 Da,旋转键数量不超过7个,XlogP小于或等于3.5。
- 移除了含有带电原子或除了碳(C)、氮(N)、硫(S)、氧(O)、氟(F)、氯(Cl)、溴(Br)、氢(H)之外的原子,或环长度超过8个原子的分子。分子还通过了药物化学筛选(MCFs)和PAINS筛选。
- 该数据集包含1,936,962个分子结构。为了进行实验,我们将数据集分为训练集、测试集和骨架测试集,分别包含约160万个、17.6万个和17.6万个分子。
- 所谓 Bemis-Murcko 骨架是去掉官能团等,去除分支,保留环包括芳香环饱和环等,保留连接键作为化学骨架的主链,提取出基础框架;
- 所谓先导分子 (Lead Compound) 是药物发现中初步筛选后,对目标生物靶标(如酶、受体或其他分子)具有活性并且潜力较高的化学物质,比如阿司匹林 (Aspirin) 的先导分子是天然化合物水杨酸(Salicylic acid)。
SMILES,SPLIT
CCCS(=O)c1ccc2[nH]c(=NC(=O)OC)[nH]c2c1,train
CC(C)(C)C(=O)C(Oc1ccc(Cl)cc1)n1ccnc1,train
CC1C2CCC(C2)C1CN(CCO)C(=O)c1ccc(Cl)cc1,test
Cc1c(Cl)cccc1Nc1ncccc1C(=O)OCC(O)CO,train
Cn1cnc2c1c(=O)n(CC(O)CO)c(=O)n2C,train
CC1Oc2ccc(Cl)cc2N(CC(O)CO)C1=O,train
O=C(C1CCCCC1)N1CC(=O)N2CCCc3ccccc3C2C1,test_scaffolds
DFT
开始提到过,原子结合成分子实际上是电子云重叠,原子轨道相互结合的结果。
简单说 DFT (密度泛函理论, Density Functional Theory) 生成分子的原理是通过求解电子密度来得到分子的稳定状态,并进一步预测其化学和物理性质。
- DFT 的目标是找到分子总能量最小化的稳定状态,即找到基态的电子密度,
- “泛函”是一个把电子密度映射到能量上的函数,根据电子密度告诉我们分子总能量。
大概跟 nn 的优化类似,比如——
- 随机初始化一个 input (电子云密度),
- weights (泛函) 是确定的 Kohn-Sham 方程,这个方程不需要优化参数,只需要迭代,
- 目标是通过改变 input 的原子坐标,改变 input 的电子云密度,以最小化 loss (分子能量)。
泛函就是函数的函数,普通的函数输入是 scalar 输出是 scalar,泛函输入是函数输出是 scalar。比如有一个温度分布 $f(x)$,泛函 $F(f)$ 可能是温度分布的总和、平均值、能量等,或比如通过积分来压缩函数信息 $F(f) = \iint_{D} f(x, y) \, dx \, dy$
3D datasets
- GEOM
- Geometric Ensemble Of Molecules,数据在 Harvard Dataverse 里。
- 37 M 分子构象,450 K 个分子,用能量和统计量标注,其中 317 K 是理化生实验性数据,133 K 来自 QM9。
- SMILES和标注之外有一个 pickle 序列化数据,用 pickle 读取后是跟上面 ZINC 一样的原子坐标数据。
- 上面是 summary.json,
- 下面是同一个分子的 pickle,包括 39 个构象 conformers。
"COCC1CCN(C(C)C(=O)NC(Cc2cc(F)cc(F)c2)C(O)C[NH2+]Cc2cccc(OC)c2)C1=O": {
"pickle_path": "rd_mols/AAAGFCUDAXPDAA-WPSXGZRWNA-O.pickle",
"datasets": [
"bace"
],
"bace": {
"class": 0.0
},
"charge": 1,
"temperature": 298.15,
"totalconfs": 34,
"uniqueconfs": 34,
"ensembleGsolv": 0.0,
"ensembleenergy": 0.8293786453390717,
"ensembleGmRRHO": 0.5259982,
"ensemblefreeenergy": 0.647714789834022,
"poplowestpct": 14.64,
"lowestenergy": -1790.2116692,
"lowestfreeenergy": -1789.6853815,
"lowestGsolv": 0.0
},
{'conformers':
[
{'rd_mol': <rdkit.Chem.rdchem.Mol object at 0x102a21b30>, 'geom_id': 191755154, 'ExTB': -114.5215816, 'Gtot': -1789.685381829991, 'homo': -5.81134502543315, 'lumo': -1.49017707949315, 'part': 2, 'Gsolv': 0.0, 'GmRRHO': 0.5262877, 'confnum': 39, 'imgfreq': 0, 'deltaExTB': 0.14966089649667502, 'deltaGtot': 0.0, 'gcpenergy': 0.050944951, 'scfenergy': -1790.2238525, 'multipoles': [{'X': 13.320834477108601, 'Y': -5.436236001799401, 'Z': 12.805190419428502}], 'totalenergy': -1790.211669529991, 'atomiccharges': {'chelpg': [-0.384107, -0.128012, -0.119892, 0.012918, -0.348897, 0.003971, -0.099417, 0.22832, -0.620608, 0.464031, -0.579857, -0.666551, 0.70218, -0.565307, 0.53173, -0.728704, 0.576836, -0.237119, -0.586949, 0.514835, -0.22303, -0.617046, -0.074913, -0.669473, -0.294498, 0.163227, -0.393489, 0.336125, -0.430043, -0.002394, -0.447307, 0.493919, -0.34994, -0.154772, -0.445046, 0.392509, -0.615911, 0.159517, 0.160476, 0.140683, 0.122743, 0.090592, 0.102362, 0.133326, 0.147082, 0.09422, 0.042913, 0.092729, 0.196191, 0.19269, 0.163939, 0.310529, 0.010566, 0.157477, 0.168965, 0.326704, 0.271008, 0.257133, 0.159122, 0.483004, 0.180651, 0.190366, 0.273248, 0.224669, 0.177406, 0.191023, 0.198102, 0.156906, 0.210417, 0.148076, 0.113059, 0.098193, 0.216593], 'lowdin': [-0.079183, -0.816131, 0.038287, -0.116671, -0.224488, 8.8e-05, -0.797974, 0.092744, -0.351816, 0.376512, -0.54649, -0.585718, 0.070386, -0.217474, -0.058601, -0.163355, 0.217519, -0.288981, -0.154311, 0.222021, -0.292465, -0.16142, 0.1198, -0.589261, 0.000245, -0.584898, 0.008839, -0.083127, -0.178494, -0.153136, -0.139364, 0.175758, -0.687728, -0.054253, -0.150924, 0.354333, -0.509984, 0.168522, 0.178438, 0.16578, 0.181807, 0.180614, 0.195278, 0.172587, 0.177462, 0.186995, 0.183336, 0.23472, 0.176017, 0.175266, 0.170432, 0.304486, 0.220894, 0.188031, 0.195249, 0.199422, 0.206502, 0.198593, 0.199265, 0.290755, 0.214907, 0.211322, 0.310346, 0.315253, 0.210142, 0.209117, 0.184257, 0.181314, 0.191326, 0.186433, 0.174786, 0.173853, 0.196207], 'mulliken': [-0.33693, -0.256748, -0.294745, 0.234326, -0.463492, -0.224222, 0.12083, -0.216166, -0.549971, 0.283058, -0.52329, 0.065622, -0.378607, -0.446257, 0.217793, -0.3163, 0.346635, -0.236717, -0.380578, 0.37968, -0.23705, -0.359167, -0.00936, -0.466132, -0.31224, -0.241289, -0.253706, 0.05608, -0.211705, -0.163229, -0.306699, 0.310531, -0.209426, -0.339675, -0.286686, 0.051327, -0.478489, 0.156809, 0.167621, 0.16549, 0.164922, 0.168774, 0.190009, 0.183647, 0.179029, 0.167752, 0.170588, 0.231494, 0.179978, 0.18388, 0.181521, 0.294058, 0.246738, 0.200581, 0.20499, 0.192331, 0.205063, 0.190918, 0.193903, 0.348514, 0.217127, 0.207198, 0.379381, 0.408151, 0.203561, 0.214364, 0.17676, 0.17631, 0.183222, 0.173553, 0.171895, 0.171776, 0.181088]}, 'boltzmannweight': 0.1463940268992422, 'dispersionenergy': -0.038761983, 'electricdipolemomentnorm': 19.260586561982183, 'deltaGxTB': None},
{'rd_mol':,'geom_id':,'ExTB':,'Gtot':,'homo':,'lumo':,'part':,'Gsolv':,'GmRRHO':,'confnum':,'imgfreq':,'deltaExTB':,'deltaGtot':,'gcpenergy':,'scfenergy':,'multipoles'': [{'X':,'Y':,'Z':}],'totalenergy':,'atomiccharges': {'chelpg': [], 'lowdin': [], 'mulliken': []}, 'boltzmannweight':,'dispersionenergy':,'electricdipolemomentnorm':,'deltaGxTB':},
···
],
'smiles': 'COCC1CCN(C(C)C(=O)NC(Cc2cc(F)cc(F)c2)C(O)C[NH2+]Cc2cccc(OC)c2)C1=O', 'datasets': ['bace'], 'bace': {'class': 0.0}, 'charge': 1, 'temperature': 298.15, 'totalconfs': 38, 'uniqueconfs': 38, 'ensembleGsolv': 0.0, 'ensembleenergy': 0.8294144900204856, 'ensembleGmRRHO': 0.5259981729924965, 'ensemblefreeenergy': 0.6477336869578462, 'poplowestpct': 14.63940268992422, 'lowestenergy': -1790.211669529991, 'lowestfreeenergy': -1789.685381829991, 'lowestGsolv': 0.0}
- ISO17
- isomers 同分异构体-17 同样来自 QM9 数据集,数据在 Quantum Machine 官网,
- 是固定原子组合 C7O2H10 不同排列的有效化学结构,有 129 个分子,每个包含 5000 种构象几何 geometries 、能量 energies 、作用力 forces 数据。
- “分子动力学轨迹的时间分辨率为1飞秒。” 数据是 .db 格式需要 SQL 操作,可以用
from ase.db import connect读写,with connect(path_to_db) as conn:,数据包含 total energy 和 atomic forces 列。Atoms(symbols='C3OC4OH10', pbc=False, cell=[1.0, 1.0, 1.0])
-11506.376711958
[[0.0, 0.0, 0.0], ...]
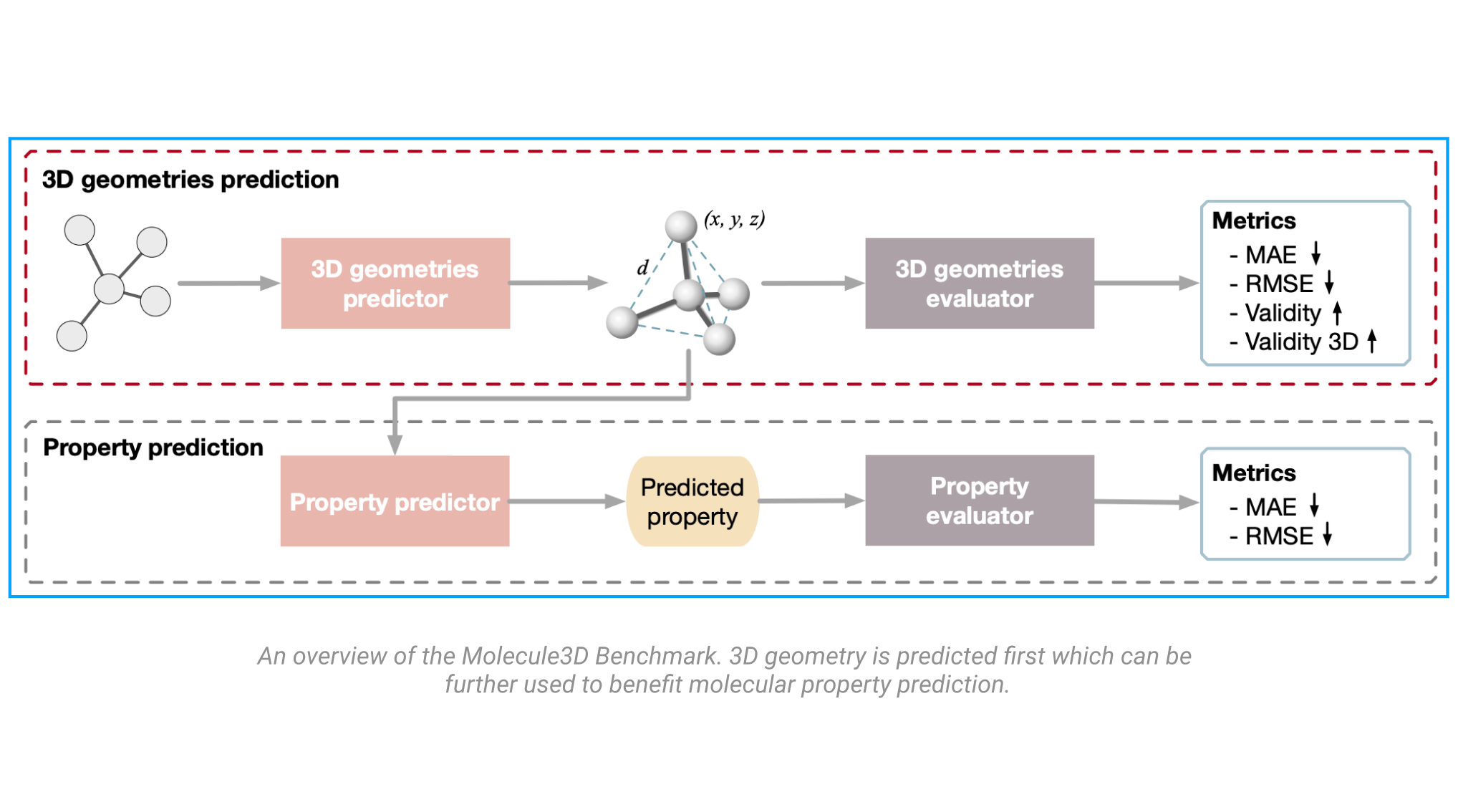
Molecule3D
4M 个从 density functional theory (DFT) 得到的分子,还提供了数据预处理、分割、训练和评价代码。数据在 Google Drive,paperswithcode 有这个 benchmark,先 3D 结构预测后性质预测。
下面 sdf 格式样本里第一行的 PUBCHEM 000000004 表示这个数据来自 PubChem,ID 第 4,B3LYP 是一种 DFT 方法,6-31G(d) 是用于计算分子轨道的函数,GAMESS 是几何优化软件,最后表示用 OpenBabel 转化数据格式;
- 第二行的 14 13 表示这个分子有 14 个原子,13 条键,V2000 是个可以用 OpenBabel 处理的数据格式;
- 之后首先是 14 个原子在当前 Å 尺度 resolution 下的坐标;
- 之后是 13 个键的连接关系, 1 2 1 表示原子 1 和 原子 2 中有个 1 键,如果是双键就是 1 2 2;
- 最后 M END 表示这个分子结束,和 dollar 结束符。
- sdf 格式数据之外有 properties.csv,列对应关系见下文例子或附录。
PUBCHEM 000000004 B3LYP 6-31G(d) gamess geometry optimization
OpenBabel05211418503D
14 13 0 0 1 0 0 0 0 0999 V2000
0.8709 -0.0593 -0.0932 C 0 0 0 0 0 0 0 0 0 0 0 0
2.3923 -0.0939 0.0366 C 0 0 1 0 0 0 0 0 0 0 0 0
2.8675 -0.1496 1.4905 C 0 0 0 0 0 0 0 0 0 0 0 0
4.3253 -0.0567 1.5320 N 0 0 0 0 0 0 0 0 0 0 0 0
2.9524 -1.1865 -0.7083 O 0 0 0 0 0 0 0 0 0 0 0 0
0.4511 0.8328 0.3857 H 0 0 0 0 0 0 0 0 0 0 0 0
0.4145 -0.9383 0.3830 H 0 0 0 0 0 0 0 0 0 0 0 0
0.5813 -0.0547 -1.1486 H 0 0 0 0 0 0 0 0 0 0 0 0
2.8285 0.7976 -0.4266 H 0 0 0 0 0 0 0 0 0 0 0 0
2.4511 -1.0709 1.9489 H 0 0 0 0 0 0 0 0 0 0 0 0
2.4412 0.6964 2.0457 H 0 0 0 0 0 0 0 0 0 0 0 0
4.7035 -0.7783 0.9198 H 0 0 0 0 0 0 0 0 0 0 0 0
4.6675 -0.2503 2.4708 H 0 0 0 0 0 0 0 0 0 0 0 0
2.5111 -1.9958 -0.4005 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0 0
1 7 1 0 0 0 0
1 6 1 0 0 0 0
2 9 1 6 0 0 0
2 3 1 0 0 0 0
3 4 1 0 0 0 0
3 10 1 0 0 0 0
3 11 1 0 0 0 0
4 13 1 0 0 0 0
5 14 1 0 0 0 0
5 2 1 0 0 0 0
8 1 1 0 0 0 0
12 4 1 0 0 0 0
M END
$$$$
cid,dipole x,dipole y,dipole z,homo,lumo,homolumogap,scf energy
000000000,4.404345,3.934717,-1.135217,-6.17698440635,-2.097997787355,4.078986618995,-20690.251813900697
000000004,-1.452589,-1.389503,1.434038,-6.1878689603699994,1.8476530448950002,8.035522005265,-6794.53585995115
- CrossDocked2020
- 数据也在 GitHub 可以获取,90 G 左右。
- label = 1 对应 RMSD(根均方偏差, 见下文 metrics)和晶体构象的偏差 ≤ 2, label = 0 是 RMSD > 2;
- 接下来的性质参数是 pK 值,若配体亲和力数据未知,则pK 值为 0;
- receptor 和 ligand 就是蛋白质受体和配体分子,对应一条数据;
- RMSD to crystal 列在 DenseNet 里要移除掉。
<label> <pK> <RMSD to crystal> <Receptor> <Ligand> # <Autodock Vina score>
1 0.0000 1.0247 1A02_HUMAN_25_199_pep_0/2gj6_D_rec_0.gninatypes 1A02_HUMAN_25_199_pep_0/2gj6_D_rec_2gj6_3ib_lig_tt_min_0.gninatypes #-3.5844
- PDB (scPDB, Binding MOAD)
- Protein Data Bank,官网说是生物学和医学领域的第一个
- PDB 有超过1TB的蛋白质、DNA和RNA的结构数据,且每年增长接近10%。
- 类似 NCBI 的 GenBank,这些数据集包罗万象,任何格式任何形式的几乎任何蛋白质和基因数据都能找到。
- DUD-E
- DUD (A Directory of Useful Decoys),decoys 是诱饵,跟真正具有好性质的目标化合物很像,但并不具有某些特性,DUD 2,950 种活性化合物,针对 40 个靶标,每个活性化合物有 36 个“诱饵”,它们的 MW 和 logP 相似,分子结构不同。
- DUD-E 是 DUD 的 Enhanced 和重建版本
- 有 22,886 个活性化合物,和它们亲和的 102 个靶标,平均每个目标靶标有 224 个配体 (ligands);
- 活性化合物每个有 50 个诱饵 decoys,有相似的物理化学属性但是不同的 2-D topology 结构。
- DUD-E 把活性化合物数据按 102 个靶标分开,比如 AA2AR 这个靶标 (target),
- 首先对应一个 PDB 条目 和对应物质量等;
- 然后对应几个活性化合物的数据文件,ism 里包括 SMILES 和 ChEMBL id 如下所示,以及 mol2 / sdf 格式的 3D 坐标数据;
- 最后有一个 receptor.pdb 文件描述靶标 (target) 分子原子构成。
| # | Target Name | PDB | Description | Method | Substances | Clustered |
|---|---|---|---|---|---|---|
| 1 | AA2AR | 3eml | Adenosine A2a receptor | By Hand | 3057 | 482 |
| 2 | ABL1 | 2hzi | Tyrosine-protein kinase ABL | Auto | 409 | 182 |
AA2AR actives_final.ism: (对应有一个decoys_final.ism存诱饵分子) |

Metrics
- 这些既可以作为模型的评价指标,也可以作为 reward function 为模型引入 reinforcement learning 思想,作为 guide gradient。
- 比如一种常用的 reward function:
- 加上penalty是:
- 分子的定量性质、特征,还会被称为分子描述符 (Descriptors),比如 RDKit 的 SAScore 函数名就是 rdMolDescriptors。
Stability
Atom Stability(原子稳定性)
定义:原子稳定性原子是否跟其他原子形成稳定的化学键,避免连接到不稳定的键上。Molecule Stability(分子稳定性)
定义:分子稳定性衡量整个分子在环境中的化学稳定性,评估分子是否容易分解、氧化或发生其他不希望的化学反应,从而确保分子在目标环境中能够长时间存在,尤其是药物分子需要在体内保持活性。
有人用 spearman’s ρ 衡量 stability。
$\rho = 1 - \frac{6 \sum_{i=1}^{n} d_i^2}{n(n^2 - 1)}$
$ d_i $ 是每对预测标签和真实标签之间的排名差异,$n$ 是样本数量,
- $\rho$:Spearman’s ρ,表示排名相关系数,取值范围从 -1 到 1。
- $\rho = 1$ 表示完全正相关(排名完全一致)。
- $\rho = -1$ 表示完全负相关(排名完全相反)。
- $\rho = 0$ 表示无相关(排名无序)。
reference 2 的 5.4 3D Molecule Evaluation 这句话是错的:
当一个原子与其他原子连接的键数与该原子的价电子数相匹配时,该原子是稳定的;当分子中所有原子都稳定时,整个分子也是稳定的。
可以参照下面 Validity 计算 Stability,我认为这两个指标可以一起算,就算原子化学键跟原子价匹配也未必稳定。
Validity
有效性:
- 基本的化学结构规则,寻找不合理的键;
- C=C=C (C₃H₄),虽然不违反化合价规则,但只能是不稳定的短暂中间体;
- C-C≡N 也是。
- 每个原子的键数是否符合其化学价 / 化合价 valence。
$\text{Validity} = \frac{\text{ # valid molecules}}{\text{ # generated molecules}}$
可以用 rdkit.Chem.Descriptors / rdMolDescriptors 计算。
Uniqueness
唯一性:希望生成的分子少重复。
$\text{Uniqueness} = \frac{\text{ # unique molecules}}{\text{ # generated molecules}}$
- $R_{\text{uniqueness}}$ 表示分子生成集合 $G$ 中的唯一性评分。
- $| \text{set}(G) |$ 是集合 $G$ 中唯一分子的数量(去除重复分子)。
- $| G |$ 是生成的分子总数。
Novelty
新颖性:希望生成训练集里不存在的分子。
$\text{Novelty} = \frac{\text{ # novel molecules}}{\text{ # generated molecules}}$
- $R_{\text{novelty}}$ 是新颖性评分。
- $x$ 表示生成的分子。
- 如果生成的分子 $x$ 不在训练集 $\text{Training Set}$ 中,则赋值为 1,表示生成了一个全新的分子。
- 如果 $x$ 在训练集中,则赋值为 0.3,表示该分子并不新颖,可能是已知的分子。
或者用 $r_{\text{novel}} = 1 - \frac{|G \cap T|}{|T|}$ ,生成的新分子也不在 T 测试集里的比例,衡量 novelty。
QED
Quantitative Estimation of Drug-likeness, 药物相似性定量估计,表示其作为药物候选分子的潜力。
Bickerton et al., 2012 的计算步骤如下:
- 数据集
- SMILES 或 InChI 格式药物分子化学结构;
- 生物活性,通常用 IC50 或 EC50;
- 用 RDKit 或 Open Babel 计算分子描述符
- 芳香环数(Aromatic Rings):表示分子中芳香环的数量。芳香环的数量可能与分子的稳定性和相互作用特性相关。
- 重原子数(Heavy Atoms):通常指除了氢以外的元素,反映了分子的复杂性。
- 环系数(Ring Count):分子中环结构的数量。环结构增加了分子的稳定性和生物活性。
- PSA
- Lipinski’s Rule of Five
- 模型训练交叉验证
- 用传统模型,比如 Linear Regression / SVM, SVR / Random Forest
- loss, 模型评估用 MSE,决定系数(R²)
R² 决定系数 (R-squared, R2) ,是衡量回归模型拟合效果的一个指标,0~1 越大越好。
AutoDock Vina
定义:AutoDock Vina是分子对接工具,用于评估分子与靶标蛋白质之间的结合亲和力,优化分子构象与受体结合能,以帮助选择结合能力最强的分子。
公式:通过评估结合模式的能量(静电、范德瓦尔斯力、疏水效应、分子旋转自由度等)预测分子之间的结合亲和力:
- $E_{\text{affinity}}$ 是分子与靶标蛋白质结合的总亲和能量,越低表示分子之间亲和力越强。
- $E_{\text{vdW}}$ 是范德瓦尔斯力,反映分子之间的非键合相互作用。
- $E_{\text{elec}}$ 是静电力,表示带电分子之间的相互作用。
- $E_{\text{hydrophobic}}$ 是疏水效应,考虑分子中的疏水基团与水分子之间的相互作用。
- $E_{\text{torsion}}$ 是分子旋转自由度的能量,反映分子旋转自由度对结合的影响。
High Affinity Percentage
定义:高亲和力百分比衡量生成的分子中具有较高结合亲和力(低结合能)分子所占的比例,用于评估生成分子是否具有良好的药物结合能力。
公式:
- $\text{Number of high affinity molecules}$ 是具有高亲和力(低结合能)的分子数量。比如用 IC50 的阈值可以设为 < 100 nM 为高亲和力,用 AutoDock Vina 的阈值可以设为 < -7 kcal/mol 结合能是高亲和力。
- $\text{Total number of generated molecules}$ 是生成的分子总数。
Synthesizability, SAScore
定义:Synthetic Accessibility Score 评估分子的合成难易度,计算多个结构特征(如键类型、环系统等)来量化分子在实际合成过程中可能遇到的难度。
在 RDKit 中,Synthetic Accessibility score(简称 SA score)是通过特定算法评估分子合成难度的。该评分系统由 Ertl et al. (2009) 提出,
原始文章:
- $ nRingBridgeAtoms $: 环桥原子数量。
- $ nSpiroAtoms $: 螺环原子数量。
- $ nStereoCenters $: 立体中心的数量。
- $ nMacrocycles $: 宏环数量。
- $ natoms $: 分子中原子的总数。
SAScore 结果通常在 -4(最差)到 2.5(最佳)之间,被乘 -1 (越高越难以合成) 后缩放至1到10之间,以提供一个更易于解释的值。
现在 RDKit 是通过 nn 实现 fragmentScore:
其中:
- $f_1, f_2, \dots, f_n$ 是描述分子结构的特征(如分子量、旋转自由度、拓扑指数等),这些特征与分子活性有一定相关性。
- $w_1, w_2, \dots, w_n$ 是对应特征的权重,通常通过回归分析或者机器学习方法来确定。
除了会考虑环结构和官能团、拓扑结构这些 penalty 之外,实现时还可能参考合成路径数据库 (Synthetic Route Database) 检查是否有已知的合成方法。
from rdkit import Chem |
Diversity
基本思路是用数据的两两相似度衡量多样性,相似度均值越低多样性越高,文章中有两种大差不差的算法,但 reference 1 的 3.2.1 Diversity & novelty Eqn. 26 公式都写错了,所以取 reference 2 equation 16 的写法。
如果多样性过高,可以通过在 reward 里增加 penalty 限制。
Coverage
其中,$S_g$ 和 $S_r$ 分别表示生成的和参考的构象,$delta$ 是给定的RMSD阈值。
Matching
定义:匹配度度量生成的分子与目标分子或参考数据库中分子之间的相似性,通过计算分子指纹的相似度来评估。
公式:
- $r_{\text{SNN}}$ 是匹配度评分。
- $G$ 是生成的分子集,$D$ 是参考数据库或目标分子集。
- $D(x_G, x_D)$ 是分子 $x_G$ 与 $x_D$ 之间的相似度。
其中,$S_g$ 和 $S_r$ 分别表示生成的和参考的构象。
1D2D ↓
KL散度
Kullback–Leibler Divergence (KLD),
KL散度是非对称的,
举个简单例子,$P=[0.4,0.6], Q=[0.5,0.5]$:
MMD
Maximum Mean Discrepancy (MMD, 最大均值差异),
假设有两个样本集 $X = \{x_1, x_2, \dots, x_n\}$ 和 $Y = \{y_1, y_2, \dots, y_m\}$,分别来自两个分布 $P$ 和 $Q$。
MMD的计算公式为:
其中,$k(x, y)$ 是核函数,通常选择高斯核(RBF核)来度量数据点之间的相似度。核函数的选择影响了MMD的表现和计算效率。
MMD依赖于一个核函数 $k(x, y)$ 来计算样本点间的相似性。常用的核函数包括:
- 高斯核:
- 线性核:用样本相似度衡量两个分布相似度的方式跟 KL 散度有些类似,但观察公式可以发现 MMD 不需要显式计算分布。
由于 KL散度 计算的例子是离散分布,所以这里举一个连续正态分布的例子说明这一点: - 分布P:一个标准正态分布 $ P \sim \mathcal{N}(0, 1) $。
- 分布Q:另一个正态分布 $ Q \sim \mathcal{N}(1, 1) $。
两个分布的差异是均值不同,一个均值为0,一个均值为1。
对于一维正态分布 $ P \sim \mathcal{N}(0, 1) $ 和 $ Q \sim \mathcal{N}(1, 1) $,我们有:
- $ p(x) = \frac{1}{\sqrt{2\pi}} \exp\left(-\frac{x^2}{2}\right) $
- $ q(x) = \frac{1}{\sqrt{2\pi}} \exp\left(-\frac{(x-1)^2}{2}\right) $
KL 散度公式:
需要确切知道 $ p(x) $ 和 $ q(x) $ 的 PDF 概率密度函数。而用 MMD 只需要从 $ P $ 和 $ Q $ 中各自抽取样本集合 $ X = \{x_1, x_2, …, x_n\} $ 和 $ Y = \{y_1, y_2, …, y_m\} $ 代入 MMD 公式:
代入核函数:
并不需要显式得到分布 PDF。
FCD
Fréchet 距离本身是计算两个路径 / 曲线的相似度,比如两个人分别在两个不同的城市出发,各自沿着某条路线行进,目标是同时到达各自的目的地,Fréchet 距离就是两个路径的“最小同时距离”,也就是“最小的最大距离”,可以想像路径越相似 Fréchet 距离值越小。
而 Fréchet ChemNet Distance (FCD) 可以衡量两个分布的距离,通常就是两个分子集合,Fréchet 距离例子里的“路径”对应分子的特征,FCD 的分子特征是 ChemNet 倒数第二层的激活值,进而计算均值 $\mu$ 和协方差 $\Sigma$ 来衡量距离。
- $\mu_1, \mu_2$ 是两个分布的均值向量。
- $\Sigma_1, \Sigma_2$ 是这两个分布的协方差矩阵。
- Tr表示矩阵的迹(即矩阵对角线元素的和)。
其中协方差公式:
3D ↓
RMSD
Root Mean Square Deviation,根均方偏差是一种衡量两个分子结构(通常是晶体构象和预测构象)之间差异的常用指标。它通过计算两组原子坐标之间的差异来量化结构的相似性。
RMSD 的值越小,表示两个结构越相似。
- 对齐两组结构:首先,需要对齐(superimpose)两个结构,以消除旋转和平移的影响,通常使用最小二乘法(least squares)来对齐。
- 计算坐标差异:对于每对对应的原子,计算它们在三维空间中的位置差异(通常使用欧几里得距离)。
- 求平方和:将每对原子之间的差异平方后加和。
- 求平均值:取所有差异平方和的平均值。
- 开平方:最后,取平均值的平方根,得到 RMSD。
公式如下:
其中:
- $N$ 是比较的原子对数。
- $r_i$ 是真实晶体结构中第 $i$ 个原子的坐标。
- $p_i$ 是预测结构中第 $i$ 个原子的坐标。
MAE
同样地,有 L2 损失就有 L1 损失,MAE (Mean Absolute Error, 平均绝对误差)。
Models
输入输出都可以是所有类型的分子表示,模型目标是按指标生成分子。
transformer + rl = Molecule generation using transformers and policy gradient reinforcement learning
Molecule Captioning
输入可以是所有类型的分子表示,输出是分子对应的自然语言描述。
- contrastive learning
- cogview, dalle, ofa
- lstm
References
三篇綜述。
- Deep learning for molecular design - a review of the state of the art, 2019, Daniel C. Elton, et al.
- MolGenSurvey: A Systematic Survey in Machine Learning Models for Molecule Design, 2022, Yuanqi Du, et al.
- A Survey of Generative AI for de novo Drug Design: New Frontiers in Molecule and Protein Generation, 2024, Xiangru Tang, et al.
Appendix
Appendix-Representations-Comparison
Appendix-QM9
| 行号 | 内容 |
|---|---|
| 1 | Number of atoms $n_a$ |
| 2 | Scalar properties (see Table 3) |
| 3,…,$na+2$ | Element type, coordinate (x, y, z, in Å), Mulliken partial charges (in e) on atoms |
| $n_{a+3}$ | Harmonic vibrational frequencies $3n_a−5$ or $3n_a-6$, in $cm^{-1}$ |
| $n_{a+4}$ | SMILES strings from GDB-17 and from B3LYP relaxation |
| $n_{a+5}$ | InChI strings for Corina and B3LYP geometries |
$n_a$=number of atoms.
Å (埃, Angstrom), $10^{-10}$ 米,0.1 纳米。会用来描述分子坐标的尺度,或者叫分辨率如 Resolution: 2.60 Å。
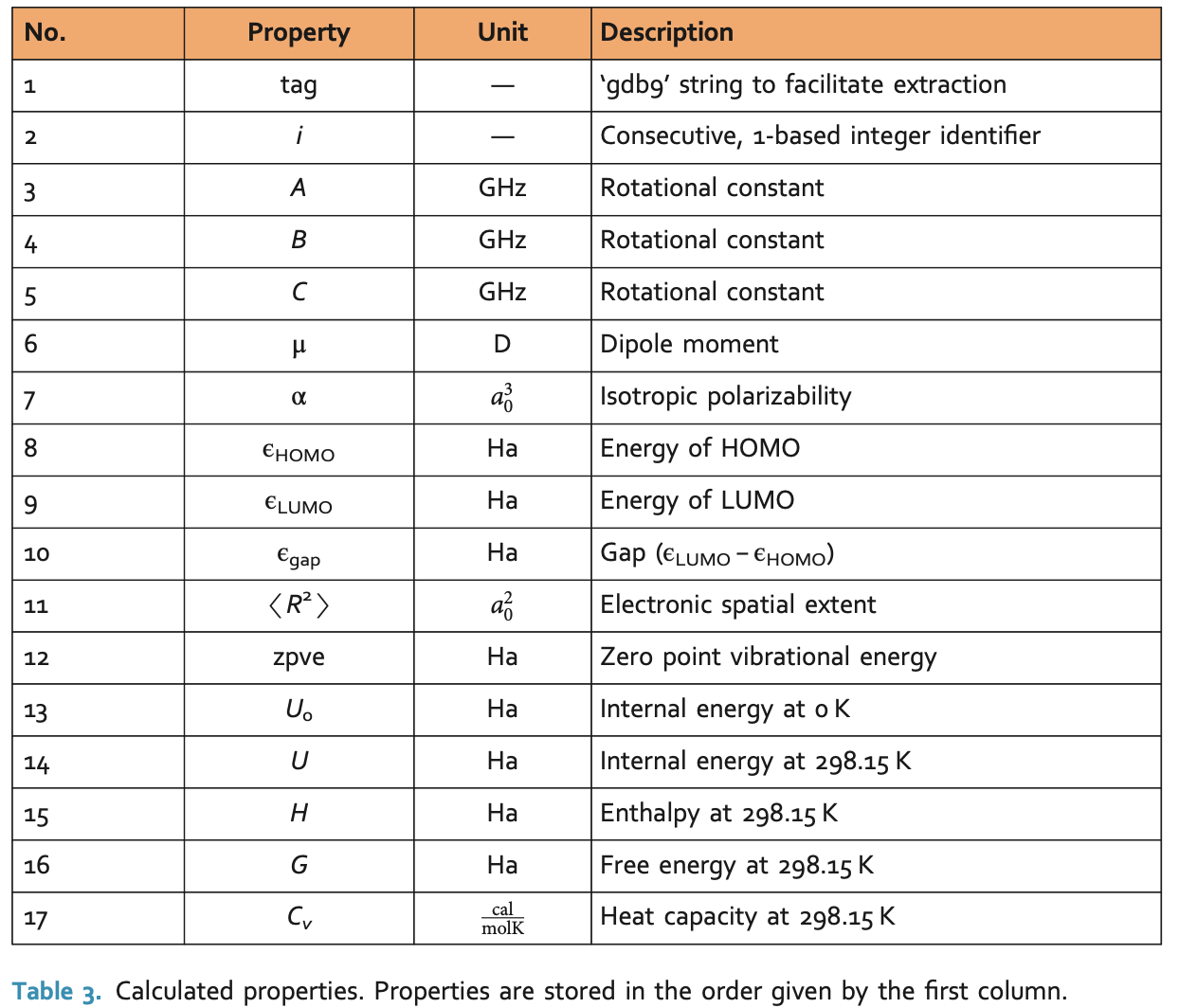
| No. | Property | Unit | Description |
|---|---|---|---|
| 1 | tag | — | ‘gdb9’ string to facilitate extraction |
| 2 | i | — | Consecutive, 1-based integer identifier |
| 3 | A | GHz | Rotational constant |
| 4 | B | GHz | Rotational constant |
| 5 | C | GHz | Rotational constant |
| 6 | μ | D | Dipole moment |
| 7 | α | $a^3_0$ | Isotropic polarizability |
| 8 | $ϵ_{HOMO}$ | Ha | Energy of HOMO |
| 9 | $ϵ_{LUMO}$ | Ha | Energy of LUMO |
| 10 | $ϵ_{gap}$ | Ha | Gap ($ϵ_{LUMO} - ϵ_{HOMO}$) |
| 11 | $⟨R^2⟩$ | $a^2_0$ | Electronic spatial extent |
| 12 | zpve | Ha | Zero point vibrational energy |
| 13 | $U_0$ | Ha | Internal energy at 0 K |
| 14 | $U$ | Ha | Internal energy at 298.15 K |
| 15 | $H$ | Ha | Enthalpy at 298.15 K |
| 16 | $G$ | Ha | Free energy at 298.15 K |
| 17 | $C_v$ | $\frac{cal}{molK}$ | Heat capacity at 298.15 K |
Properties are stored in the order given by the first column.
Appendix-ChEMBL
properties available
| 字段名称 | 描述 | 数据类型 |
|---|---|---|
| molecule_chembl_id | ChEMBL ID | 字符串 |
| pref_name | Name | 字符串 |
| molecule_synonyms | Synonyms | 对象 |
| molecule_type | Type | 字符串 |
| max_phase | Max Phase | 字符串 |
| molecule_properties.full_mwt | Molecular Weight | 双精度浮点数 |
| _metadata.related_targets.count | Targets | 整数 |
| _metadata.related_activities.count | Bioactivities | 整数 |
| molecule_properties.alogp | AlogP | 双精度浮点数 |
| molecule_properties.psa | Polar Surface Area | 双精度浮点数 |
| molecule_properties.hba | HBA | 整数 |
| molecule_properties.hbd | HBD | 整数 |
| molecule_properties.num_ro5_violations | #RO5 Violations | 整数 |
| molecule_properties.rtb | #Rotatable Bonds | 整数 |
| molecule_properties.ro3_pass | Passes Ro3 | 字符串 |
| molecule_properties.qed_weighted | QED Weighted | 双精度浮点数 |
| molecule_properties.cx_most_apka | CX Acidic pKa | 双精度浮点数 |
| molecule_properties.cx_most_bpka | CX Basic pKa | 双精度浮点数 |
| molecule_properties.cx_logp | CX LogP | 双精度浮点数 |
| molecule_properties.cx_logd | CX LogD | 双精度浮点数 |
| molecule_properties.aromatic_rings | Aromatic Rings | 整数 |
| structure_type | Structure Type | 字符串 |
| inorganic_flag | Inorganic Flag | 整数 |
| molecule_properties.heavy_atoms | Heavy Atoms | 整数 |
| molecule_properties.hba_lipinski | HBA (Lipinski) | 整数 |
| molecule_properties.hbd_lipinski | HBD (Lipinski) | 整数 |
| molecule_properties.num_lipinski_ro5_violations | #RO5 Violations (Lipinski) | 整数 |
| molecule_properties.mw_monoisotopic | Molecular Weight (Monoisotopic) | 双精度浮点数 |
| molecule_properties.np_likeness_score | Np Likeness Score | 双精度浮点数 |
| molecule_properties.molecular_species | Molecular Species | 字符串 |
| molecule_properties.full_molformula | Molecular Formula | 字符串 |
| molecule_structures.canonical_smiles | Smiles | 字符串 |
| molecule_structures.standard_inchi_key | Inchi Key | 字符串 |
| molecule_structures.standard_inchi | Inchi | 字符串 |
| withdrawn_flag | Withdrawn Flag | 布尔值 |
| orphan | Orphan | 整数 |
| _metadata.compound_records.compound_key | Records Key | 字符串 |
| _metadata.compound_records.compound_name | Records Name | 字符串 |
Appendix-Molecule3D
| Target ID Number | Property |
|---|---|
| 0 | Dipole x |
| 1 | Dipole y |
| 2 | Dipole z |
| 3 | HOMO |
| 4 | LUMO |
| 5 | HOMO-LUMO Gap |
| 6 | SCF Energy |